






《Nature》重磅:应激调控异染色质遗传,或破解肿瘤耐药难题
异染色质以组蛋白H3第9位赖氨酸甲基化(H3K9me)为标志,可通过细胞分裂进行表观遗传继承,维持基因沉默,从而保持细胞身份并使其适应环境挑战。对粟酒裂殖酵母(Schizosaccharomyces pombe)的研究表明,异染色质的传递依赖于"读-写"机制,其中组蛋白去乙酰化酶稳定化的、具有足够密度的H3K9me3修饰核小体可将Clr4SUV39H富集于染色质上,促进H3K9甲基化的进一步沉积。作为E3泛素连接酶复合物ClrC的亚基,Clr4SUV39H是否受其他机制调控以控制异染色质传递,此前尚不清楚。本研究发现了一个泛素依赖性的异染色质遗传调控枢纽(HRH),该枢纽广泛调控异染色质传递,即使在缺乏组蛋白去乙酰化酶活性的情况下亦然。HRH受到限制因子Raf1DDB2的调控,Raf1 DDB2是ClrC泛素连接酶的底物受体。除了将Clr4SUV39H连接到染色质上的其他ClrC组分外,Raf1 DDB2还以剂量依赖的方式促进组蛋白H3第14位赖氨酸(H3K14)的泛素化,这对异染色质的自我传递至关重要。HRH与环境响应通路(包括无义介导的mRNA降解(NMD)和雷帕霉素靶蛋白(TOR)信号通路)紧密相连,使细胞能够适应不断变化的环境条件。通过调控异染色质传递,细胞利用HRH获得抗真菌药物抗性并适应高温环境。总之,作者的研究示了异染色质自我传递通过H3K14泛素化受到外部刺激的主动调控,这对理解生理和疾病过程中表观遗传景观快速变化的机制具有广泛意义。该研究于2026年1月发表在《Nature》,IF 48.5分。
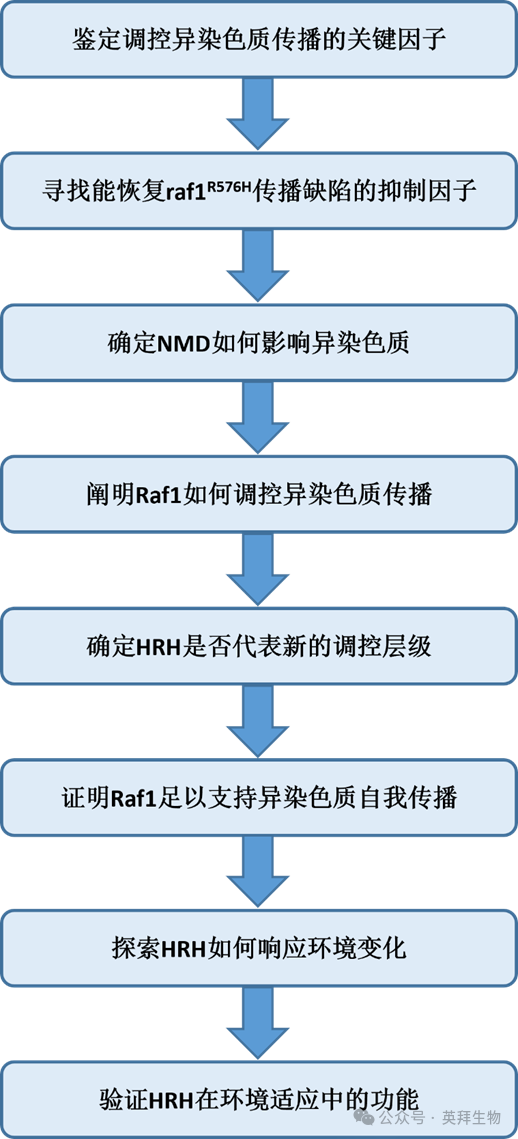
技术路线:
主要研究结果:
1、NMD影响异染色质传递
粟酒裂殖酵母ClrC与CUL4-DDB1-DDB2 E3泛素连接酶复合物具有结构相似性。除Clr4SUV39H甲基转移酶外,ClrC还包括WD-40蛋白Raf1、锌指蛋白Raf2、β-螺旋桨蛋白Rik1以及cullin家族蛋白Cul4。Rik1与人类DDB1高度相似,后者与CUL4共同参与组蛋白甲基化修饰。Raf1与人类DDB2相似,可能是一种DCAF(DDB1和CUL4相关因子),作为E3泛素连接酶的底物受体。Raf1具有两个保守的WDxR基序(第515-518位和第573-576位残基)。
为探究Raf1中WDxR基序的功能,作者构建了raf1R518H和raf1R576H等位基因,并利用敏感的REIIΔ mat2P::ura4+报告基因评估mat区域的异染色质沉默。在缺乏局部REII沉默子的mat1-M细胞中,异染色质组装缺陷导致mat2P去抑制和单倍体减数分裂,表现为细胞暴露于碘蒸气时呈深棕色染色。相比之下,具有功能性异染色质沉默的细胞呈黄色染色。附近的mat2P::ura4+报告基因在缺乏尿嘧啶(-URA)的培养基或反选择性的5-氟乳清酸(FOA)培养基上测试时,为异染色质沉默提供额外的读数。raf1R518H和raf1R576H均表现出mat2P::ura4+沉默的丧失和深色碘染色。然而,它们对cenH成核位点插入的ura4+(Kint2::ura4+)沉默的影响不同。raf1R518H中的Kint2::ura4+表现出严重的沉默缺陷,mat区域H3K9me3丢失,而raf1R576H细胞中的Kint2::ura4+仅受到轻微影响。值得注意的是,raf1R576H在cenH成核中心周围维持了H3K9me3,但未能将其扩散到周围序列。
随后作者研究了raf1R576H如何影响异染色质扩散。作者对raf1R576H细胞中恢复REIIΔ mat2P::ura4+沉默的因子进行了无偏遗传筛选(图1a)。三个抑制子重建了沉默,表现为在FOA培养基上生长增加和浅色碘染色。其中两个突变定位于esl1(也称为ebs1),一个定位于upf1(图1b)。Upf1和Esl1是NMD通路的组分。删除upf1或esl1重现了对raf1R576H沉默缺陷的抑制(图1c),并通过ura4+报告基因的反转录PCR(RT-PCR)分析得到证实(图1d)。染色质免疫沉淀测序(ChIP-seq)显示,raf1R576H upf1Δ和raf1R576H esl1Δ中沉默mat区域的H3K9me3和Swi6HP1增加(图1e,f),亚端粒区域的异染色质扩散也有类似增加。相比之下,单突变体和双突变体的着丝粒H3K9me3水平与野生型(WT)细胞相当,这与RNAi定向ClrC持续靶向近着丝粒重复序列一致。这些结果表明,NMD组分的丢失可以挽救raf1R576H的异染色质传递缺陷。
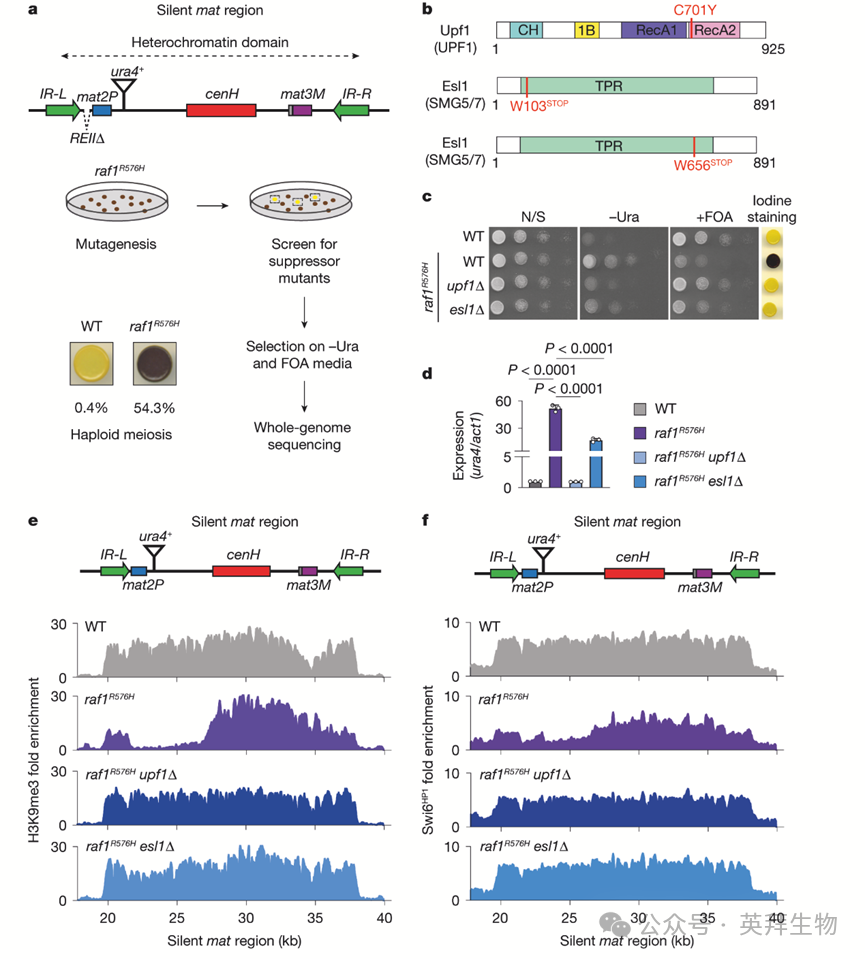
图1:NMD成分的缺失挽救了异染色质扩散缺陷
2、NMD调控ClrC亚基Raf1DDB2水平
为解释NMD缺失如何抑制raf1R576H中的异染色质缺陷,作者考虑到NMD与RNAi竞争着丝粒重复序列转录本,因此NMD缺陷会增强RNAi介导的小RNA产生和异染色质组装。然而,Upf1的丢失并未增加小RNA产生。此外,在raf1R576H upf1Δ细胞中,RNAi因子Ago1对于恢复沉默和沉默mat区域H3K9me3富集是可有可无的。此外,即使当RNAi依赖性异染色质成核中心cenH被ura4+替代(KΔ::ura4+)时,upf1Δ细胞相对于WT细胞的H3K9me3水平和异染色质沉默也有所增加。因此,NMD组分丢失对raf1R576H细胞中异染色质缺陷的抑制并非由于RNAi介导的异染色质组装增加。
作者探究NMD是否调控异染色质传递所需关键因子的表达。RNA测序(RNA-seq)显示,与WT相比,raf1R576H中raf1 mRNA水平降低(图2a)。raf1R576H upf1Δ细胞中raf1 mRNA水平升高,超过WT水平(图2a)。这种效应是raf1特异性的,因为其他ClrC组分编码的转录本未受影响。RNA免疫沉淀(RIP)显示Upf1与raf1 mRNA结合,提示NMD直接控制raf1表达(图2b)。事实上,NMD通路缺陷增加了raf1 mRNA和Raf1蛋白水平(图2c)。Upf1的丢失导致raf1表达升高,不仅在raf1R576H中如此,在raf1WT细胞中也是如此。因此,尽管作者使用低表达raf1R576H突变等位基因进行的遗传筛选对于鉴定raf1作为NMD靶标至关重要,但这些分析表明NMD也抑制raf1WT表达。因此,raf1是一个参与调控异染色质传递的真正NMD靶标。
作者注意到raf1 mRNA含有与RNA降解相关的隐蔽性、低效剪接内含子。这些内含子在upf1Δ细胞中被检测到,与靶RNA降解通路被抑制时隐蔽内含子的出现一致。作者的分析还揭示,与NMD类似,剪接机器也影响raf1 mRNA。从最初涉及NMD参与raf1调控的筛选中(图1a),作者鉴定了两个额外的突变体:一个携带upf2突变(upf2也在NMD中发挥作用),另一个携带sap49突变(sap49编码U2 snRNP相关RNA结合剪接因子)。在raf1R576H细胞中引入sap49A175V突变恢复了沉默mat区域的异染色质沉默和H3K9me3扩散,并部分恢复了亚端粒的H3K9me3扩散。在RNAi持续靶向异染色质的着丝粒处,H3K9me3未观察到重大变化。sap49A175V显著上调raf1 mRNA和Raf1蛋白,与upf1Δ相似。结合剪接与NMD之间的先前联系,这些结果表明Sap49可能通过NMD促进raf1 mRNA降解。
3、Raf1驱动异染色质传递
为确定NMD丢失是否仅通过增加Raf1水平来恢复异染色质传递,作者在诱导型启动子下过表达raf1R576H(raf1R576H-oe),并作为对照过表达raf1WT(raf1-oe)。表达水平与upf1Δ细胞中相当的Raf1恢复了raf1R576H细胞中的沉默(扩展数据图3h,i)。raf1R576H-oe对沉默的挽救程度与raf1WT-oe相同(图2d)。沉默的恢复伴随着沉默mat区域H3K9me3的扩散(图2e),且raf1R576H-oe与raf1WT-oe细胞中的H3K9me3水平相当。这些发现表明R576H突变并未损害Raf1功能;相反,raf1R576H细胞中的异染色质缺陷反映了Raf1丰度的降低。raf1R576H-oe还挽救了raf1R576H细胞中亚端粒异染色质扩散缺陷(图2f)。因此,NMD介导的Raf1丰度调控是异染色质传递调控中的关键控制点。
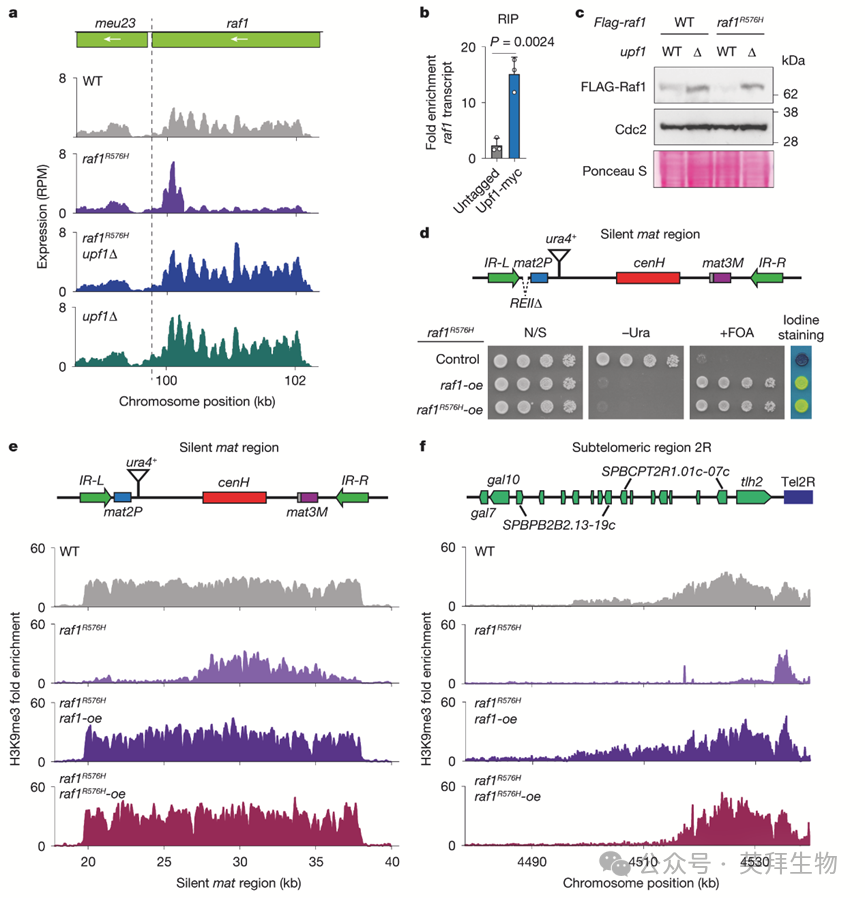
图2:NMD机制调节ClrC亚基Raf1DDB2的表达
4、Raf1DDB2丰度调控ClrC结合
随后作者研究了Raf1丰度如何影响H3K9me3和异染色质传递。作者的分析表明,Raf1是ClrC组装的限制因子,特别是在招募Clr4SUV39H方面(及其他功能,见下文)。甘油梯度分级分离显示,相当一部分Clr4SUV39H以单体形式存在,表明其与其他ClrC组分的结合较弱(图3a)。Raf1过表达时,Clr4SUV39H向更高分子量组分迁移(图3a)。这种迁移与Clr4SUV39H掺入ClrC的增加相关,在raf1-oe和upf1Δ细胞中均观察到Clr4SUV39H与ClrC亚基Raf2的结合增强(图3b)。因此,Raf1水平可能调控Clr4SUV39H与其他ClrC组分的结合。
Raf1 WD重复结构域形成β-螺旋桨折叠,介导蛋白质相互作用。作者探究Raf1是否直接与Clr4SUV39H结合。Raf1 WD重复区域在体外结合Clr4SUV39H(扩展数据图4b)。在体内,Clr4SUV39H与Raf2的结合与Raf1丰度相关。缺乏Raf1的细胞中Clr4无法与Raf2结合,而在Raf1水平较低的raf1R576H细胞中仅检测到弱结合(图3c)。相比之下,携带raf1WT等位基因的细胞中,随着Raf1水平升高,Clr4-Raf2结合增加,Raf1过表达显著增强了Clr4与Raf2的结合。这些发现进一步表明,Raf1是Clr4SUV39H掺入ClrC的限制因子。
为确定Raf1水平是否影响ClrC的染色质结合状态,作者在活细胞中追踪Raf2的动态。HaloTag标记的Raf2(Raf2-Halo)形成两到三个焦点,对应于核周聚集的异染色质位点。单分子追踪(SMT)揭示了慢速和快速扩散的Raf2-Halo颗粒及其扩散系数(D)。作为染色质结合对照,Halo标记的组蛋白H2B(H2B-Halo)显示出两种相似的扩散状态。Raf1过表达时,慢扩散Raf2群体增加,表明ClrC重新分布到移动性较低的染色质结合状态。此外,存活分布显示Raf1过表达时染色质结合ClrC的解离减少。
为进一步分析ClrC移动性,作者使用系统级分类算法扰动-期望最大化(pEM)将Raf2轨迹分类为不同的扩散状态。鉴定出三种亚扩散Raf2移动状态。状态3的约束半径(Rc)对应于平均核半径,与H2B未观察到的自由状态一致。状态1和2具有较小的Rc值,被认为是受限状态。值得注意的是,Raf2-Halo受限状态1和2的D和Rc与两种H2B状态相似,表明它们是染色质结合状态。Raf1过表达导致Raf2受限状态1和2的比例增加,自由状态减少,表明Raf2分子向染色质相关群体转移。总之,这些结果表明,较高水平的Raf1不仅促进ClrC完整性,还增加ClrC与染色质结合的比例。
Raf1水平增加时观察到的ClrC染色质结合增强也可从Raf2定位的直接可视化中得到证实。raf1-oe和upf1Δ细胞(两者均含有较高Raf1水平)中Raf2-Halo焦点的数量和强度显著增加。在raf1-oe细胞中,较高的Raf2焦点强度与Raf2扩散增强相关,特别是在缺乏明确异染色质边界的亚端粒区域,而被边界元件侧翼的异染色质区域(如沉默mat区域)的Raf2分布保持不变。Raf1过表达还增加了ClrC在异染色质岛的结合,包括对发育或环境信号响应的基因,如pho1。
总之,这些分析表明,升高的Raf1稳定并延长ClrC染色质结合,浓缩这一组蛋白甲基转移酶和泛素连接酶复合物,最终促进驱动异染色质传递的"读-写"活性。
5、Raf1DDB2 DCAF指导H3K14泛素化
ClrC在组蛋白H3第14位赖氨酸处进行单泛素化(H3K14ub),这在体外增强Clr4SUV39H甲基转移酶活性,但其体内相关性尚不清楚。作者探究组蛋白H3是否为ClrC E3连接酶的生理底物,以及Raf1是否在剂量依赖性影响ClrC染色质结合之外促进H3K14ub。
作者使用抗H3K14ub抗体通过ChIP-seq全基因组分析H3K14ub。H3K14ub在主要异染色质结构域富集,包括沉默mat区域、近着丝粒和亚端粒,但在缺乏Raf1的细胞中完全消失(图3d)。这一结果表明H3K14ub是一个显著的异染色质标志,其沉积需要DCAF Raf1。Raf1过表达显著增加H3K14ub,特别是在缺乏边界元件的亚端粒,且这种增加与H3K9me3扩散增强相关(图3e)。因此,Raf1剂量对H3K14ub和H3K9me3至关重要,仅增加Raf1即可增强异染色质传递。
为验证H3K14ub在异染色质传递中的需求,作者检测了E2泛素结合酶Ubc4突变体ubc4-1。H3K14ub在异染色质结构域(包括mat区域)显著降低(图3f)。由于Ubc4不是具有泛素化和H3K9甲基化双重功能的ClrC核心组分,该突变分离了H3K14ub对异染色质组装的贡献。在ubc4-1中,H3K14ub的丢失与沉默mat区域异染色质扩散和沉默受损相关(图3g,h)。尽管H3K9me3在RNAi机器招募ClrC的cenH成核位点建立,但在H3K14ub缺陷的ubc4-1突变体中未能扩散(图3h)。Raf1过表达未能恢复ubc4-1细胞中的H3K14ub、H3K9me3扩散或异染色质沉默(图3f,g,h)。
H3K14ub在体外刺激Clr4SUV39H甲基转移酶活性,因此预计会过度激活Clr4SUV39H,导致H3K9me3积累,从而刺激异染色质传递的"读-写"机制。相应地,ubc4-1可能破坏H3K14ub与Clr4SUV39H的串扰,将H3K9me3降低到有效异染色质传递所需阈值以下。
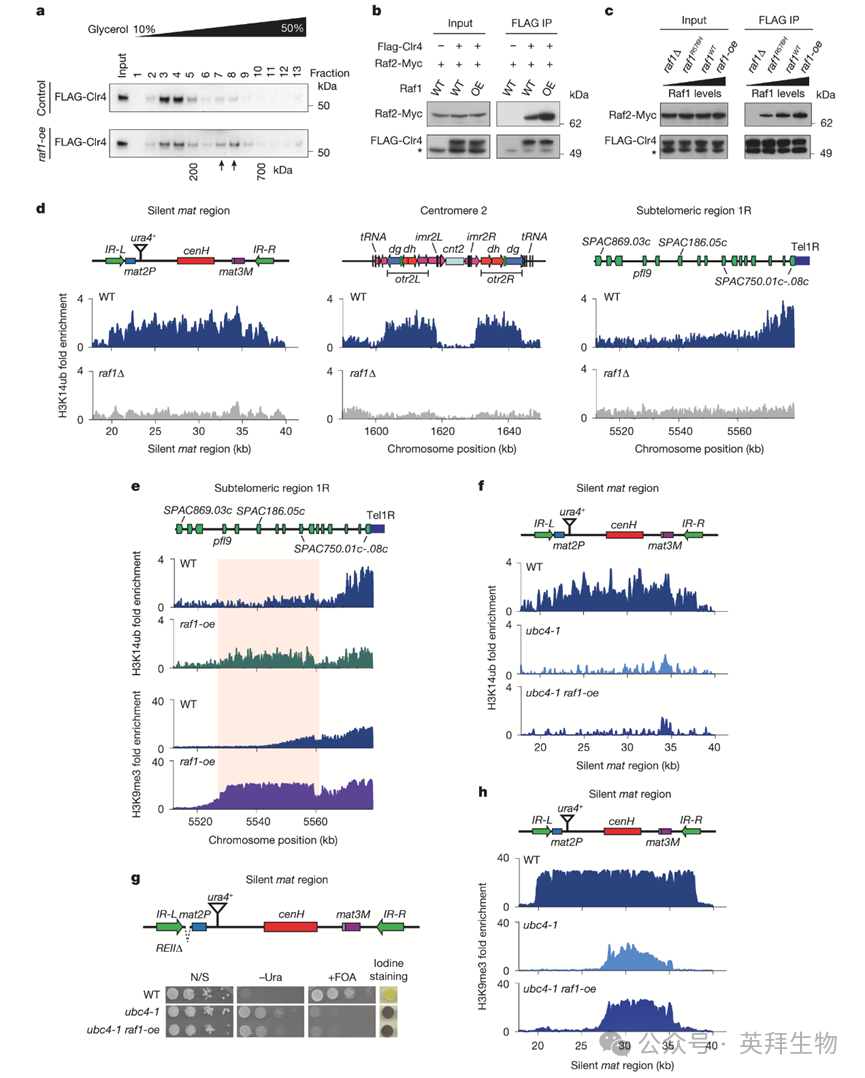
图3:Raf1DDB2连接Clr4SUV39H和ClrC,并介导H3K14ub促进异染色质扩散
6、H3K14ub定义新的调控通路
使H3K14去乙酰化的HDAC Clr3对异染色质传递至关重要。通过抑制组蛋白周转,Clr3维持Clr4"读-写"活性所需的足够H3K9me3密度。Clr3的丢失降低H3K9me3密度并损害异染色质传递和沉默(图4a)。作者还发现clr3Δ细胞中H3K14ub降低,特别是在cenH成核位点周围区域(图4b),这促使作者检测升高Raf1是否能恢复异染色质组装。
通过raf1-oe或upf1Δ增加Raf1,在clr3Δ细胞中恢复了REIIΔ mat2P::ura4+报告基因的异染色质沉默(图4a),并与沉默mat区域H3K14ub水平增加相关(图4b)。虽然clr3Δ细胞主要在cenH附近显示H3K9me3富集,但H3K9me3在clr3Δ raf1-oe和clr3Δ upf1Δ细胞中扩散至整个结构域(图4c)。这提示一种平行机制,即增强Raf1水平(增强ClrC染色质结合和H3K14ub)可绕过Clr3 HDAC活性在异染色质传递中的需求(图4d)。
clr3Δ细胞中Raf1上调表型模拟抗沉默因子Epe1的丢失。由于Epe1经历泛素依赖性蛋白降解,作者探究Raf1是否改变Epe1丰度。WT和raf1-oe细胞中Epe1-GFP(绿色荧光蛋白)焦点数量和强度相似,表明Raf1并非通过降低Epe1水平发挥作用。遗传分析显示,Epe1和Raf1独立作用以抑制Clr3催化突变体(clr3D232N)细胞的沉默缺陷。删除epe1或upf1各自部分恢复REIIΔ mat2P::GFP报告基因沉默,双缺失产生叠加效应和近乎完全的挽救。结合Raf1过表达时Epe1水平不变的结果,这些结果支持Raf1介导的调控异染色质传递的独特通路。
为进一步验证Raf1调控异染色质传递的机制,作者检测增加Raf1是否能缓解其他情境下的缺陷。在携带三个组蛋白H3拷贝之一突变(hht2G13D)的细胞中,H3K9me3密度降低损害异染色质传递。值得注意的是,在hht2G13D细胞中引入raf1-oe或upf1Δ恢复了REIIΔ mat2P::ura4+沉默。hht2G13D细胞在沉默mat区域也显示H3K14ub缺陷,该缺陷在upf1Δ细胞中被增加的Raf1水平挽救,并伴随结构域H3K9me3扩散的恢复。
作者进一步探究升高Raf1是否能绕过异染色质传递的其他需求。确实,raf1-oe或upf1Δ在缺乏FACT(促进染色质转录)亚基Pob3、SMARCAD1 SNF2重塑因子Fft3或核膜蛋白Amo1NUPL2的细胞中恢复沉默,这些因子维持H3K9me3密度。总之,这些结果鉴定了Raf1介导的ClrC染色质结合和H3K14ub调控作为支持异染色质维持和传递的先前未被认识的机制。
7、Raf1水平控制自我传递
作者检测了Raf1丰度是否影响序列非依赖性的异染色质遗传。通过将TetR-Clr4(Clr4SUV39H与TetR DNA结合结构域融合)可逆地锚定于ade6+报告基因上游的六个四环素操纵子位点,在异位位点成核异染色质(图4e)。无四环素时(tetR-clr4ON),TetR-Clr4诱导H3K9me3并使ade6+沉默(红色菌落)。释放TetR-Clr4(tetR-clr4OFF)后,H3K9me3和ade6+沉默迅速丢失,可能是因为内源性Clr4SUV39H的局部染色质浓度不足以维持"读-写"活性和异位异染色质。相比之下,在Raf1水平升高的upf1Δ细胞中释放TetR-Clr4,使异位位点的ade6+沉默和H3K9me3持续数代(图4e),表明增加Raf1丰度(增强ClrC染色质结合)足以支持异染色质自我传递。
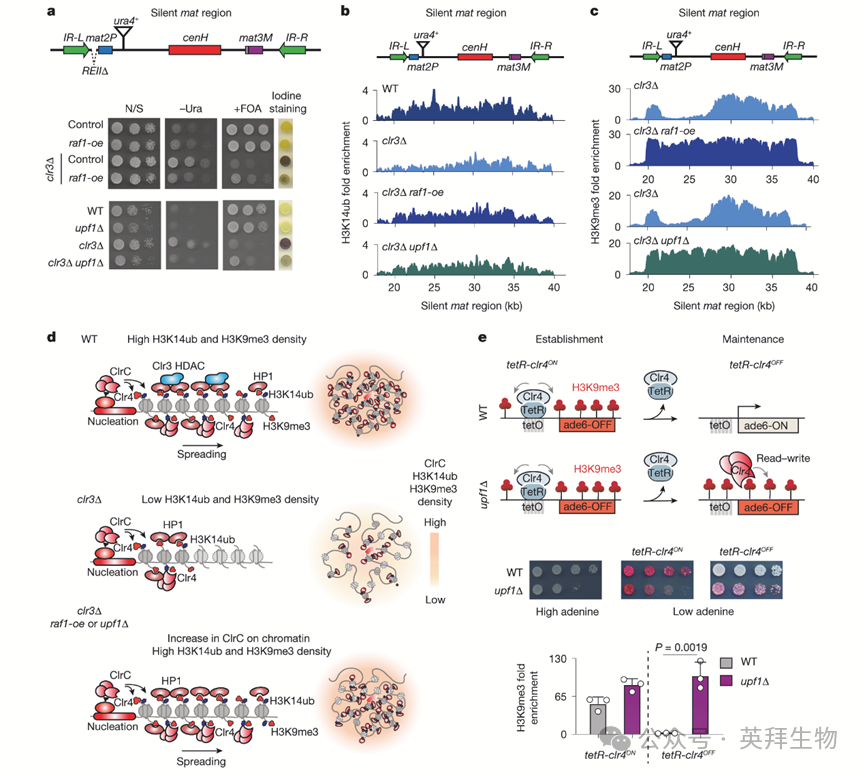
图4:Raf1DDB2dcafa -directed H3K14ub揭示了异染色质增殖的新途径
8、适应过程中Raf1丰度的调控
NMD调控的Raf1水平显著影响异染色质传递,提示该机制在生理条件下运作。异染色质的自我传递使可遗传的基因重编程和环境适应成为可能。近期研究表明,细胞通过异染色质传递获得咖啡因抗性。由于咖啡因在其他系统中减弱NMD活性,作者研究了咖啡因处理是否影响粟酒裂殖酵母的NMD。咖啡因存在时NMD调控的转录本上调。此外,raf1R576H突变转录本去抑制,与upf1Δ中类似(图2a),相应蛋白水平增加。咖啡因处理也显著增加WT Raf1水平(图5a)。
作者探究Raf1丰度单独是否有利于咖啡因抗性的发展。Raf1过表达与咖啡因抗性菌落增加相关(图5b),并对抗真菌药物氟康唑和克霉唑产生抗性(图5c)。upf1Δ细胞中观察到类似抗性(图5c)。相比之下,Raf1过表达在缺乏Clr4SUV39H的细胞中未能赋予咖啡因抗性,提示异染色质依赖性机制。raf1-oe咖啡因抗性细胞的H3K9me3 ChIP-seq显示,已知异染色质岛和新基因组位点的H3K9me3增加和扩散,包括沉默赋予咖啡因抗性的位点,如与线粒体功能障碍相关的cup1。因此,Raf1上调是增强异染色质稳定化的自然机制的一部分,使细胞能够适应不利条件。
9、TOR-Raf1节点调控异染色质
作者探索了其他条件是否影响Raf1水平。37°C培养细胞降低Raf1水平,鉴于各种系统中观察到的温度依赖性异染色质改变,这令人深思。37°C时Raf1降低导致异染色质扩散受损和基因沉默破坏(图5d,e)。Raf1过表达恢复沉默和沉默mat间隔的H3K9me3分布(图5d,e),提示细胞响应高温下调Raf1,从而影响异染色质传递。
作者接下来检测调控Raf1的信号通路。Tor1(TORC2复合物亚基)的丢失严重降低Raf1蛋白和转录本水平。相比之下,其他激酶突变(包括Tor2)不影响Raf1水平,Tor2是TORC1亚基,靶向减数分裂基因H3K9me的RNA加工机器,但对其他部位异染色质组装是可有可无的。为检测温度对Tor1活性的影响,作者检测了30°C和37°C时Gad8(人类AKT同源物)磷酸化。Gad8磷酸化在30°C时高,37°C时检测不到(图5f)。缺乏Gad8的细胞也显示Raf1降低,与tor1Δ类似。这些发现提示高温使Tor1信号级联失活,后者是适当Raf1表达所必需的。tor1Δ和gad8Δ细胞中Raf1水平降低也导致咖啡因抗性菌落减少,Raf1过表达可逆转该表型。
基于作者的结果,作者假设Raf1丰度降低驱动了tor1Δ细胞中先前报道的异染色质缺陷。确实,在upf1Δ中增加Raf1抑制了tor1Δ背景中的异染色质传递和沉默缺陷(图5g,h)。然而,upf1Δ tor1Δ双突变体中Raf1水平低于单独upf1Δ(图5i),表明Tor1独立于NMD影响Raf1。支持这一点的是,37°C培养upf1Δ细胞与30°C相比降低Raf1表达(图5i)。如预期,将upf1Δ tor1Δ细胞从30°C移至37°C未引起进一步降低(图5i)。因此,Upf1和Tor1通过不同通路控制Raf1丰度。这些发现支持一个模型,即多种输入汇聚于Raf1水平的控制,将Raf1定位为调控异染色质稳健性和可遗传性的调控枢纽的关键组分(图5j)。
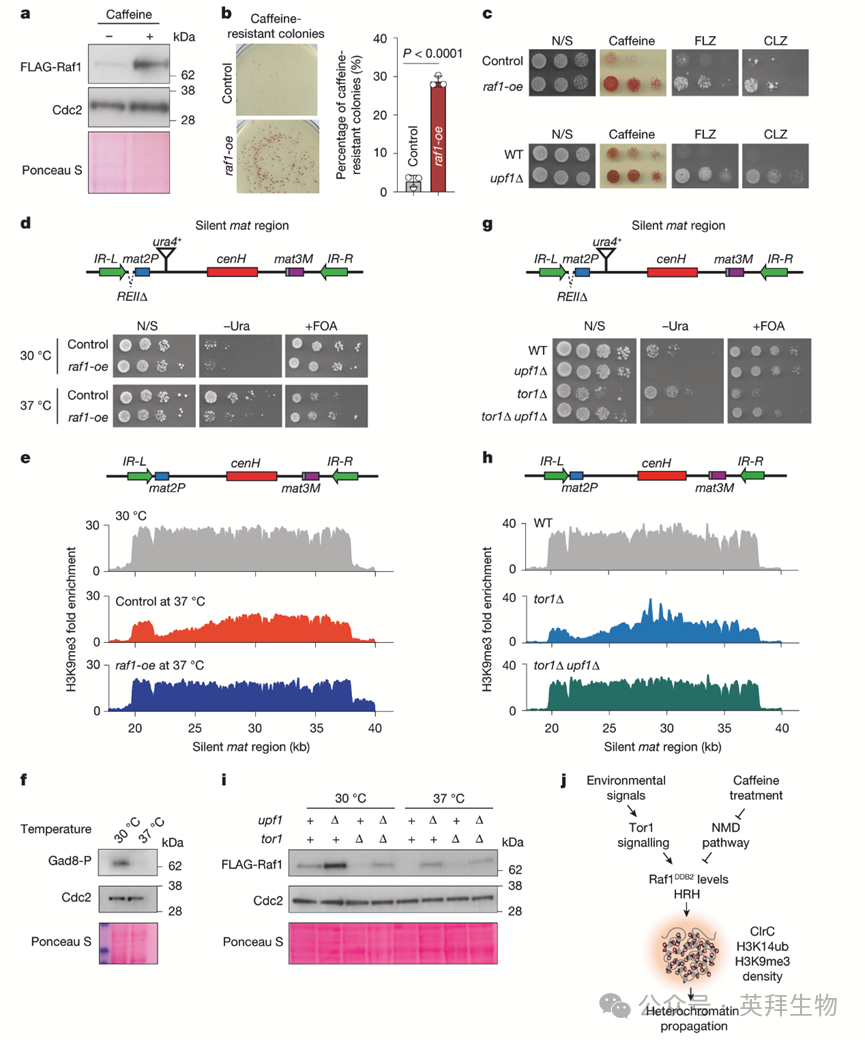
图5:在异染色质适应过程中,Raf1DDB2水平受到自然调节
10、应激下保存异染色质
鉴于基于Raf1的HRH在异染色质传递中的关键作用,作者想知道影响Raf1表达的因子是否影响异染色质的表观遗传遗传。由于高温显著降低Raf1水平,作者假设这将破坏异位异染色质结构域的自我传递。作者使用含有插入tetO-ade6+基因旁的一个或两个Clr3吸引序列(CAS)拷贝的稳健报告基因检测这一点。CAS招募Clr3 HDAC,促进有效异染色质传递。
作者在30°C培养的细胞中通过锚定TetR-Clr4(tetR-clr4ON)于CAS-tetO-ade6+报告基因建立异染色质,然后在30°C或37°C释放TetR-Clr4(tetR-clr4OFF)后评估沉默异染色质状态的传递。30°C时,异染色质状态表观遗传遗传,传递效率随CAS元件数量增加而提高,两个CAS3拷贝提供最有效的异染色质维持。相反,37°C培养的细胞未能维持沉默状态。
作者随后探究增加Raf1是否挽救高温下异染色质传递的缺陷。使用2xCAS3-tetO-ade6+报告基因,作者比较了WT和upf1Δ细胞中异位异染色质结构域的传递。释放TetR-Clr4后,upf1Δ细胞即使在37°C也传递沉默的ade6+,而WT仅在30°C维持沉默状态的遗传。ChIP显示upf1Δ比WT更大程度保存H3K9me3。
这些发现确立了不同生长条件下Raf1水平的控制是一个真正的异染色质HRH。该枢纽通过调控ClrC染色质结合和H3K14ub水平调节异染色质稳定性,并在适应环境挑战期间被利用。
结论:
该研究鉴定了一个以Raf1为限制因子的异染色质遗传调控枢纽(HRH),发现应激通过NMD和TOR信号通路调控Raf1丰度,进而通过H3K14ub依赖机制控制异染色质自我传播,使细胞能够适应环境变化并获得药物抗性。该研究揭示了环境因素通过H3K14ub调控异染色质遗传的分子机制,为理解发育、疾病及病原体抗药性中的表观遗传可塑性提供了新范式。
参考文献:
Bhatt B, Wei Y, Pradhan AK, Dhakshnamoorthy J, Zofall M, Xiao H, Vijayakumari D, Jain S, Folco HD, Qi H, Ball DA, Karpova TS, Wheeler D, Wong J, Grewal SIS. Stress controls heterochromatin inheritance via histone H3 ubiquitylation. Nature. 2026 Jan 7. doi: 10.1038/s41586-025-09899-8IF: 48.5 Q1 . Epub ahead of print. PMID: 41501458.