






破解“冷肿瘤”之谜:USP7-TREX1泛素开关重启放疗免疫协同杀伤
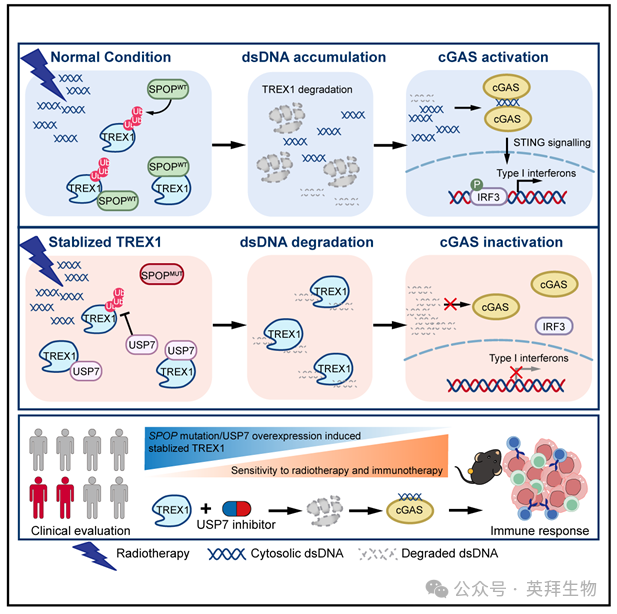
癌细胞中cGAS-STING信号的激活需要由内在或治疗诱导的DNA损伤产生的胞质DNA。然而,临床上利用这一通路改善免疫疗法的努力收效有限,DNA损伤与免疫疗法之间联系的认识仍存在空白。本研究发现,泛素化介导的胞质DNA降解是DNA损伤后cGAS-STING激活的关键决定因素。机制上,胞质DNA外切酶TREX1会被E3泛素连接酶SPOP降解,但又被去泛素化酶USP7反向稳定。癌症相关的SPOP突变或USP7过表达会提高TREX1水平,促进胞质DNA降解,削弱cGAS-STING介导的免疫激活。值得注意的是,在接受放化疗的患者中,USP7表达升高与肿瘤浸润淋巴细胞减少及疾病进展加速相关。此外,USP7抑制剂可降低TREX1水平,并在放疗后恢复免疫应答。总之,作者的这些发现阐明了DNA损伤与免疫激活之间的机制联系,并提示USP7抑制剂有望成为放射免疫疗法的增强剂。该研究于2026年1月发表在《Cancer Cell》,IF 44.5分。
技术路线:
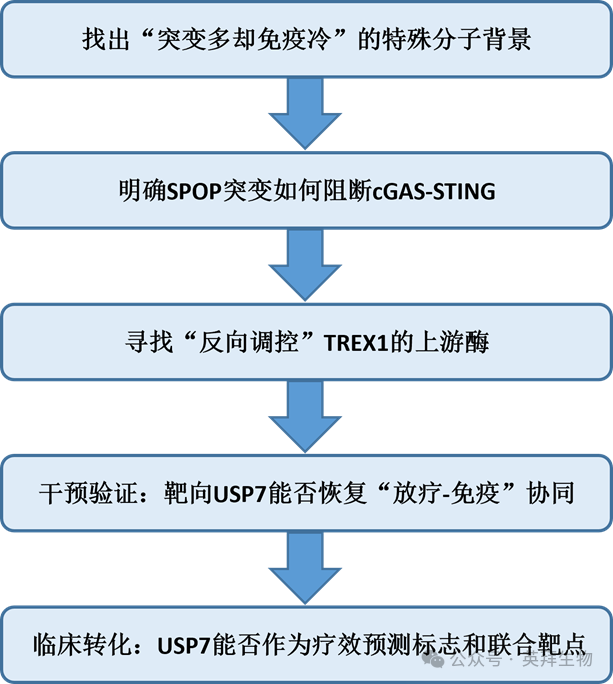
主要研究结果:
1)遗传筛选发现SPOP突变可减少DNA损伤后胞质dsDNA累积
利用TCGA泛癌数据集38与TIMER2免疫浸润数据39–41,作者发现多数HR基因突变患者TMB升高且免疫浸润增强;唯独SPOP突变在TMB升高的同时却伴随免疫浸润减少(图1A、1B)。单癌种分析进一步证实,在PRAD与UCEC中,SPOP突变虽提高TMB或新抗原数量,却未像SPOP野生型(WT)患者那样增加CD4+或CD8+ T细胞浸润(图1C、1D)。为独立验证,作者分析了本中心100例原发前列腺癌。Sanger测序检出20例携带SPOP突变。DNA损伤/基因组不稳定标志物γ-H2AX的免疫组化显示,SPOP突变前列腺癌组织γ-H2AX表达升高(图1E、1F),但CD8+ T细胞浸润却减少(图1G)。这一“悖论”现象提示,SPOP突变是研究DNA损伤与免疫应答关联的理想模型。
DNA损伤时核内dsDNA漏入胞质是免疫激活的关键触发因素。作者进一步检测最常见SPOP突变F133V对损伤后胞质dsDNA分布的影响。电离辐射(IR)可诱导胞质dsDNA出现,而稳定表达SPOP-F133V突变体显著抑制PC-3和C4-2前列腺癌细胞中胞质dsDNA的累积(图1H、1I);SPOP敲除细胞亦得类似结果。胞质dsDNA定量试剂盒显示,SPOP突变或敲除加速损伤后胞质dsDNA的衰减(图1J)。组分分离实验证实,SPOP突变或敲除减少损伤后>20 bp的胞质dsDNA片段累积(图1K)。综上,SPOP突变前列腺癌细胞在DNA损伤后降低胞质dsDNA累积,可能借此逃避免疫激活。
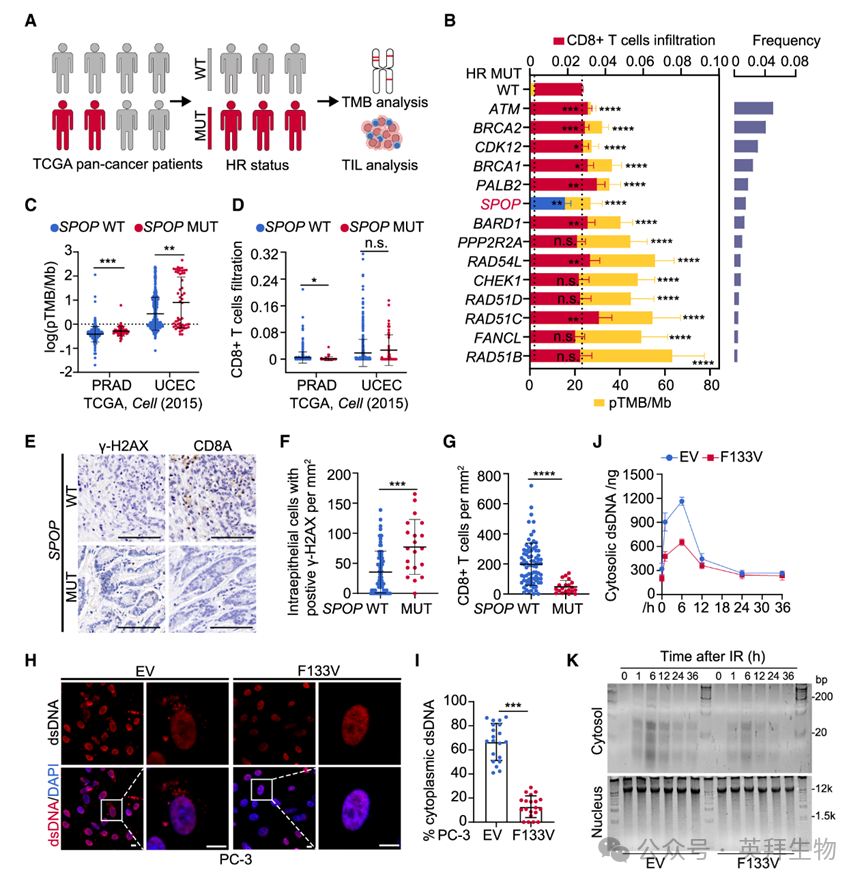
图1:遗传筛选发现SPOP突变减少DNA损伤后胞质dsDNA累积
2)SPOP突变无法启动DNA损伤后的cGAS-STING免疫应答
胞质dsDNA经cGAS-STING通路感知是先天免疫的核心。为探究SPOP突变细胞中胞质dsDNA减少是否影响该通路,作者在IR后检测其激活情况。PC-3与C4-2对照细胞IR后p-STING、p-IRF3水平升高,I型干扰素应答基因(IFNB1、IFIT1、CXCL10)表达及cGAMP水平增加;而SPOP突变或敲除细胞无上述反应(图2A–2C)。同样,IR诱导的cGAS胞质分布及微核形成在SPOP突变/敲除细胞中被废除(图2D、2E)。临床相关模型进一步验证:来自SPOP-WT或F133L突变前列腺癌灶的PDX模型中,IR未改变新抗原数量;IR后SPOP-F133L PDX肿瘤p-IRF3未升高,而WT组升高,且WT肿瘤ISG表达上调,突变组无变化(图2G、2H)。本中心患者肿瘤组织亦显示,SPOP突变肿瘤p-IRF3水平低于WT组(图2I、2J)。TCGA前列腺癌/子宫内膜癌数据集44同样表明,SPOP突变与WT肿瘤间干扰素应答无差异(图2K)。综上,SPOP突变削弱DNA损伤后的cGAS-STING免疫激活。
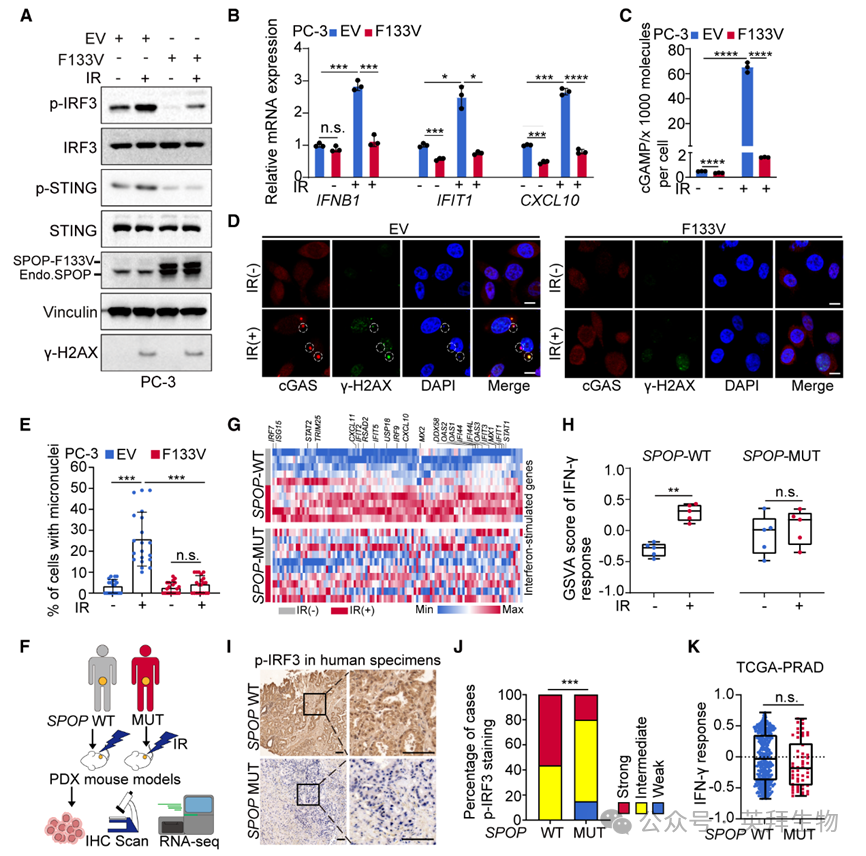
图2:SPOP突变无法启动DNA损伤后的cGAS-STING免疫应答
3)TREX1被SPOP泛素化并降解
为探究SPOP突变如何影响胞质dsDNA累积,作者检测了SPOP与多种已知外切酶的相互作用。结果发现,SPOP仅特异性结合TREX1,而不与其他外切酶互作。值得注意的是,IR处理后短时间内,SPOP突变PC-3和C4-2细胞中TREX1蛋白水平持续升高。进一步实验显示,外源SPOP以剂量依赖方式降低TREX1蛋白,该效应可被蛋白酶体抑制剂MG132完全阻断;相反,SPOP敲除升高内源TREX1蛋白,但不影响其mRNA(图3A、3B)。重新引入SPOP可恢复TREX1降解,并增强IR后胞质dsDNA累积及I型干扰素应答(图3C、3D)。上述结果提示TREX1是SPOP的泛素化底物。确证实验表明,SPOP促进TREX1多聚泛素化,缩短其半衰期;缺失SPOP则呈相反效应,且均可被SPOP回补逆转(图3E–3G)。为排除其他SPOP底物(如BRD4)的干扰,作者用BRD4抑制剂JQ1处理PC-3和C4-2细胞,结果JQ1对TREX1蛋白水平及DNA损伤后cGAS激活均无影响。SPOP靶蛋白通常含保守结合基序SBC:[AVP]. [ST][ST][ST];TREX1 C端247AVTTT251即符合该基序(图3H)。删除这5个氨基酸后,SPOP在293T细胞中完全失去与TREX1互作、泛素化及降解的能力(图3I、3J),证实247AVTTT251为功能性SBC。功能上,TREX1缺失增强IR后cGAS-STING激活(p-STING、p-IRF3升高)、胞质dsDNA累积及I型干扰素应答;而重新表达缺失SBC的TREX1突变体对上述指标的逆转作用强于野生型TREX1(图3K–3M)。综上,TREX1是SPOP的新底物,SPOP通过泛素化降解TREX1调控胞质dsDNA累积。
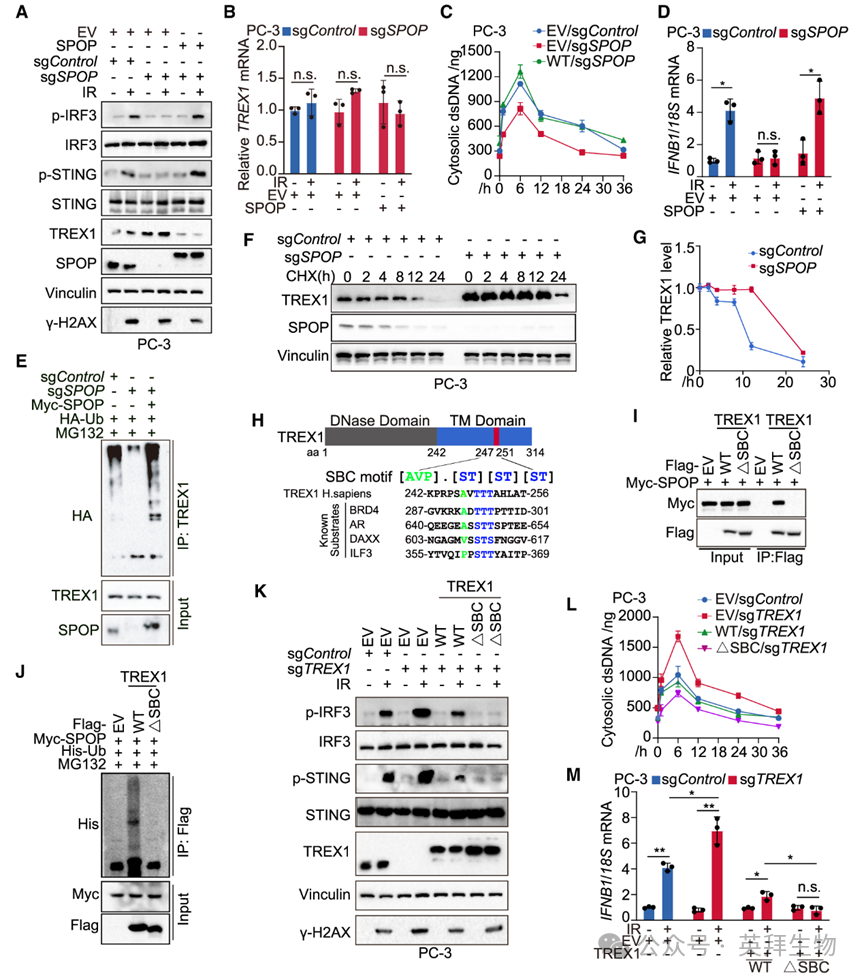
图3:TREX1被SPOP泛素化并降解
4)病理性SPOP突变削弱其降解TREX1及调控胞质dsDNA的能力
前列腺癌中SPOP突变集中于负责底物结合的MATH结构域。Co-IP显示,SPOP ΔMATH突变体丧失与TREX1结合能力;ΔBTB突变体缺失CUL3结合位点,无法泛素化底物,但仍能结合TREX1,却不能促进其泛素化。作者进一步构建4种前列腺癌相关及4种子宫内膜癌相关SPOP突变体,Co-IP表明这些突变体与TREX1结合能力均弱于野生型(图4A),并显著降低SPOP介导的TREX1泛素化、延长其半衰期(图4B–4D)。功能上,TREX1敲除增加IR后胞质dsDNA累积及干扰素应答;而在PC-3细胞中,过表达野生型或突变型SPOP均不影响胞质dsDNA水平及干扰素应答(图4E、4F)。临床样本分析显示,75%的SPOP突变肿瘤呈TREX1强阳性,而SPOP野生型肿瘤仅20%强阳性,50%为中等阳性(图4G、4H)。结果表明,病理性SPOP突变破坏其降解TREX1的能力,从而削弱DNA损伤后胞质dsDNA的调控。
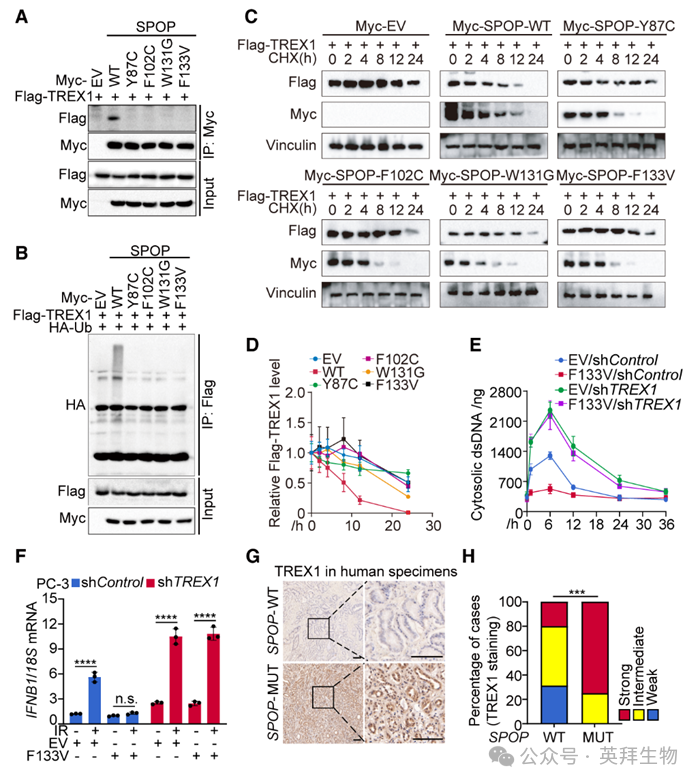
图4:病理性SPOP突变削弱其降解TREX1及调控胞质dsDNA的能力
5)去泛素化酶USP7与TREX1互作并阻止其蛋白酶体降解
尽管SPOP突变肿瘤中TREX1蛋白水平升高,但在部分SPOP野生型肿瘤中TREX1总蛋白仍维持高位(图4G、4H),提示去泛素化酶等其他机制可能参与调控TREX1表达。为此,作者用12种靶向15个去泛素化酶的商品化抑制剂处理PC-3细胞,发现仅USP7抑制剂XL177A可降低TREX1蛋白(图5A、5B)。RNA-seq显示,在众多去泛素化酶抑制剂中,XL177A特异性增强干扰素γ及α应答(图5C、5D)。XL177A呈剂量依赖性降低TREX1蛋白,但不影响其mRNA;该效应可被蛋白酶体抑制剂MG132逆转(图5E),提示XL177A通过蛋白酶体途径减少TREX1。FT671和GNE-6776两种USP7抑制剂同样提高TREX1泛素化水平、缩短其半衰期。内源性Co-IP证实PC-3细胞中USP7与TREX1互作。野生型USP7而非催化失活突变体C223A可减少TREX1多聚泛素化并延长其半衰期。体外泛素化实验显示,SPOP催化TREX1多聚泛素化,而USP7共存显著抑制该过程(图5F)。USP7敲除亦增加TREX1泛素化、缩短其半衰期,表型与XL177A一致(图5G、5H、S10E–S10G)。临床样本中USP7与TREX1表达正相关(图5I、5J)。TCGA泛癌数据多变量分析表明,USP7表达是PRAD队列免疫浸润的独立预测因子,并与多种癌种CD8+ T细胞浸润独立相关(图5K)。综上,USP7是调控TREX1去泛素化、维持其稳定性的关键去泛素化酶。
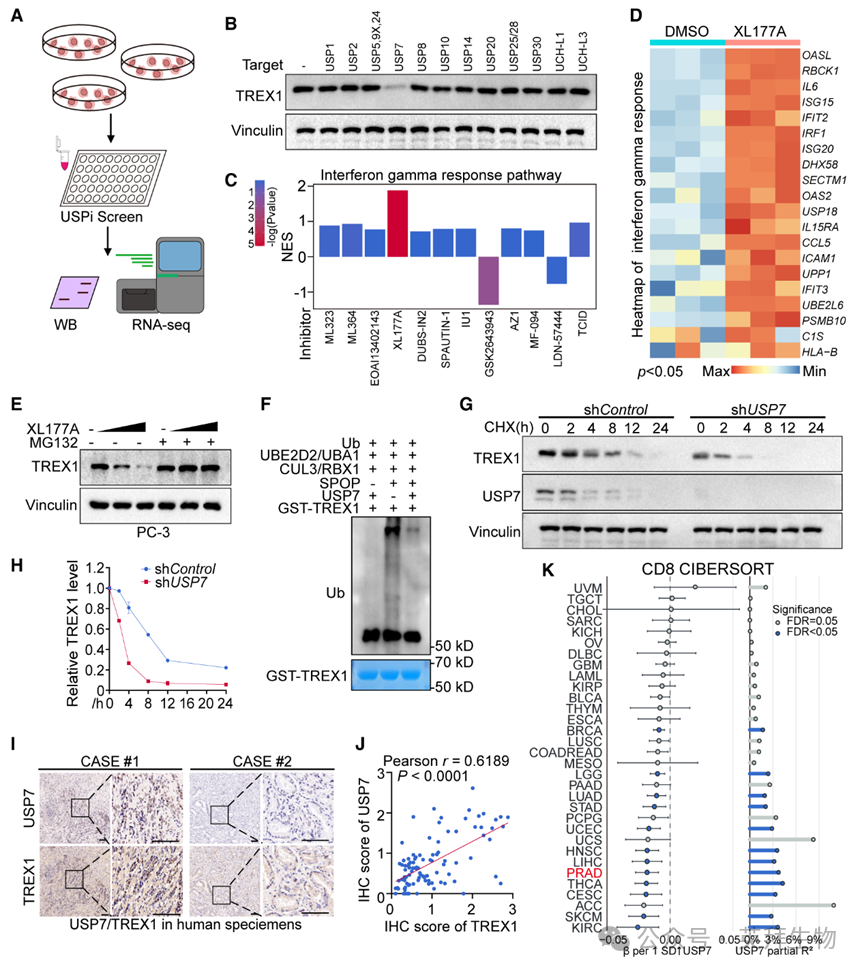
图5:去泛素化酶USP7与TREX1互作并阻断其蛋白酶体降解
6)USP7抑制促进DNA损伤后胞质dsDNA累积及cGAS-STING激活
与上述结论一致,USP7敲除提高IR后PC-3细胞胞质dsDNA水平(图S11A、S11B),并抑制损伤后胞质dsDNA衰减(图6A、6B)。IR后USP7敲除细胞中STING、IRF3磷酸化升高,cGAMP水平及IFNB1表达增加,微核形成与cGAS激活亦增强(图6C–6G)。在SPOP突变前列腺癌模型中,单独突变无法触发损伤后胞质dsDNA累积或cGAS-STING激活,而XL177A处理可逆转该缺陷,恢复WT及F133V突变PC-3、C4-2细胞的胞质dsDNA累积与通路激活(图6H–6K)。FT671与GNE-6776亦在IR后成功激活PC-3、C4-2细胞的cGAS-STING通路。因此,无论SPOP状态如何,USP7抑制均可增强DNA损伤后的胞质dsDNA累积与cGAS激活。
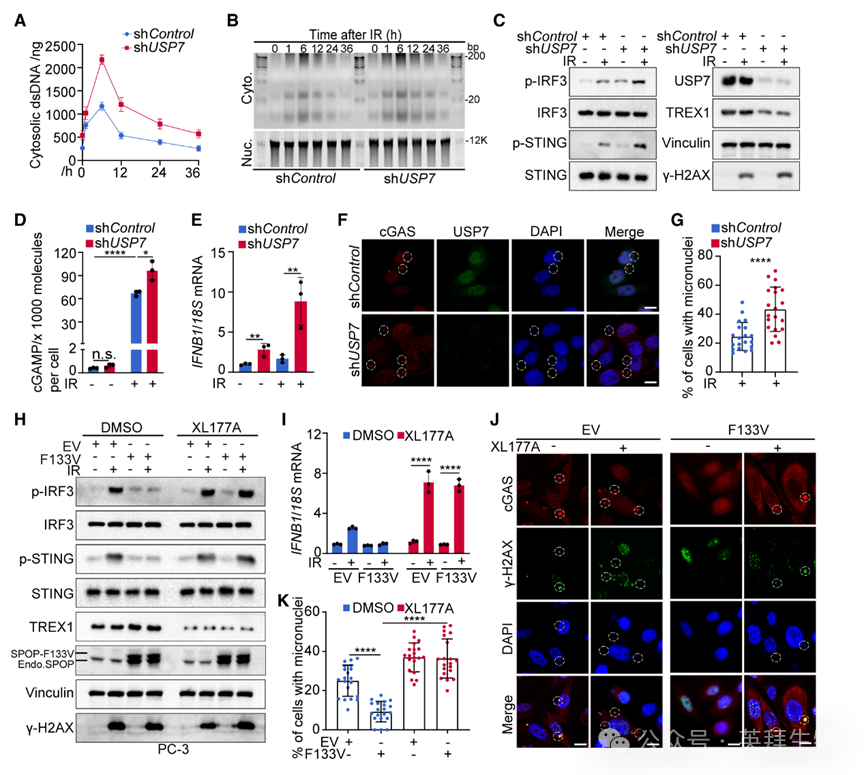
图6:USP7抑制促进DNA损伤后胞质dsDNA累积并激活cGAS-STING
7)临床证据:USP7介导放化疗后免疫抑制
为验证USP7-TREX1轴在临床免疫抑制中的作用,作者分析了42例接受同步放化疗+免疫治疗的晚期宫颈癌患者系列标本(原前列腺癌队列获取治疗后样本困难)。SiHa与C-33A宫颈癌细胞系证实SPOP-USP7-TREX1调控关系与前列腺癌一致。治疗前后宫颈活检构建组织芯片,配对血清用于肿瘤标志物分析(图7A)。多重免疫组化显示USP7与TREX1蛋白水平正相关(图7B、7C)。低USP7肿瘤在放化疗后可见cGAS-STING激活及CD3+/CD4+/CD8+ TIL浸润;高USP7肿瘤则显著抑制IRF3磷酸化及TIL浸润(图7D–7F)。血清学分析显示,USP7高表达与治疗后SCC、CEA、CA125水平升高相关,提示疗效差(图7G、7H)。临床随访表明,USP7高表达预测2年内疾病快速进展(图7I)。因此,USP7既是放化疗+免疫治疗耐药的预后生物标志,也是潜在治疗靶点。MD安德森食管鳞癌样本亦显示低USP7与CD8A高表达相关(图7J)。但在STING缺失SW49及cGAS缺失A375细胞中,USP7敲除无法激活IR后cGAS-STING通路,证实USP7介导的免疫抑制依赖完整的cGAS-STING信号轴。
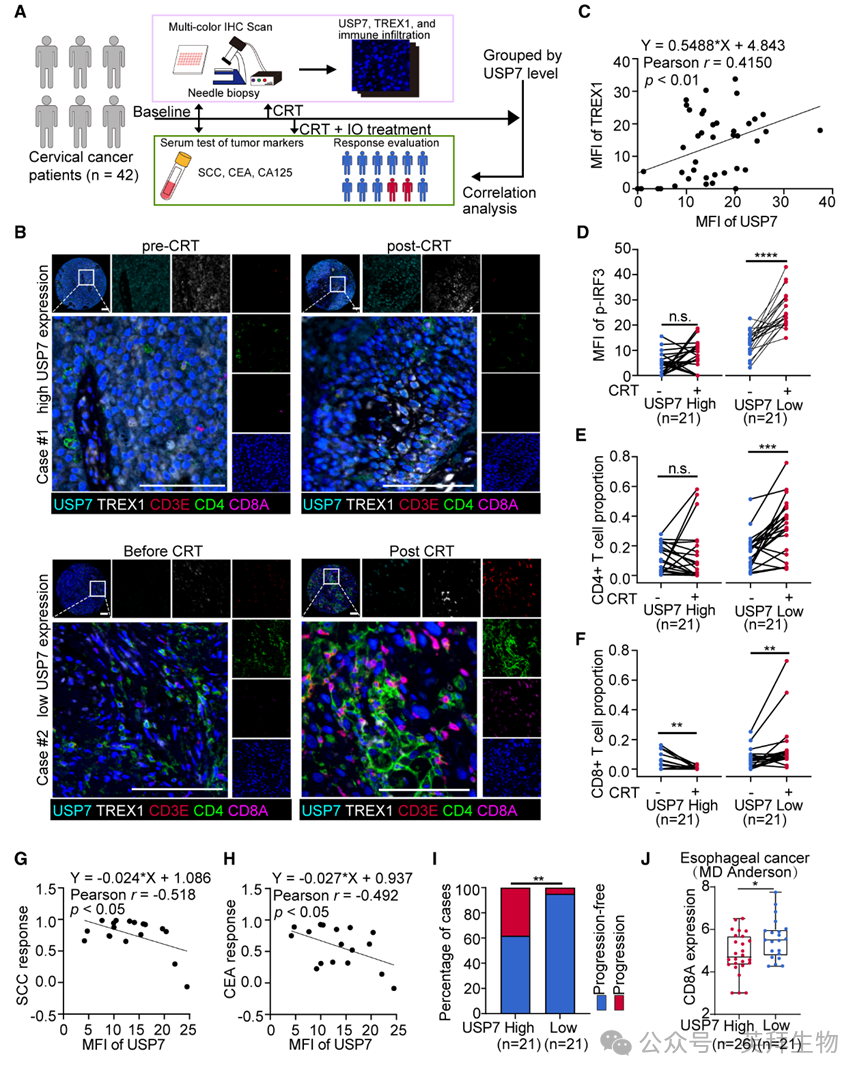
图7:USP7介导放化疗后免疫抑制的临床证据
8)靶向USP7可增强前列腺癌放疗与免疫治疗疗效
基于USP7通过抑制胞质dsDNA累积及cGAS-STING激活削弱抗癌免疫的发现,作者推测USP7抑制剂可与放疗及免疫治疗协同。为此,作者在同源RM1移植瘤小鼠(Usp7野生型或敲低)中联合应用放疗+免疫治疗(图8A)。结果显示,IR+抗CTLA4抗体组10只小鼠中3只存活;额外敲低Usp7则减缓肿瘤生长,存活率升至6/10(图8B、8C)。机制上,Usp7敲低显著降低TREX1蛋白,升高IRF3磷酸化,并增加联合治疗组CD3+、CD4+及CD8+ TIL(图8D、8E)。USP7抑制剂XL177A在RM1模型中获得类似疗效与生存获益(图8F–8J)。将TREX1 ΔSBC突变体重新导入Trex1敲除RM1细胞后,XL177A+放化疗组中,携带ΔSBC突变的小鼠肿瘤生长更快、生存更差,而野生型TREX1组则相反;ΔSBC突变对IR诱导的cGAS-STING激活及TIL浸润的抑制也强于野生型,证实TREX1是SPOP下游效应因子。为模拟晚期人类前列腺癌,作者建立Pten/Rb1双敲(DK)小鼠前列腺癌细胞系(C57BL/6背景),该模型呈现低分化肿瘤且免疫“冷”微环境。GNE-6776联合放化疗同样抑制Pten/Rb1 DK肿瘤生长并延长生存。安全性评估中,C57BL/6J小鼠接受3倍于有效剂量的XL177A后,心、肝、脾、肺、肾H&E染色未见明显组织损伤;血清生化显示,免疫相关炎症因子轻度升高,但肝肾功能仍在正常范围。综上,USP7抑制可安全有效地增强前列腺癌放疗与免疫治疗的疗效。
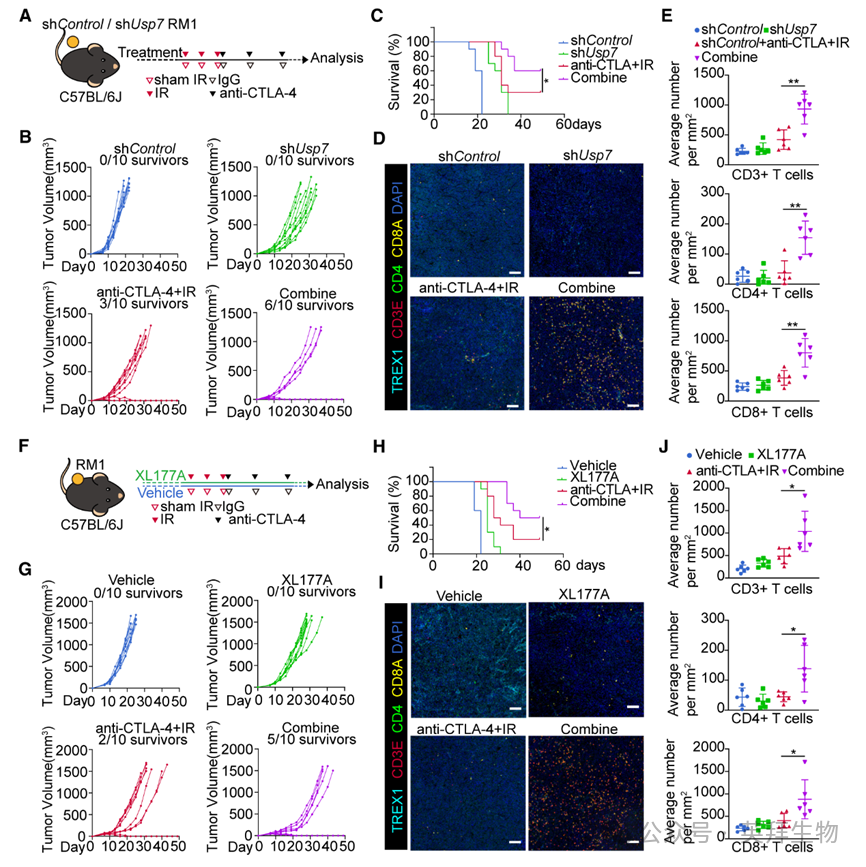
图8:靶向USP7增强前列腺癌放疗与免疫治疗的疗效
结论:
综上所述,作者的研究发现,SPOP突变和USP7过表达共同导致TREX1上调,引发胞质DNA过度降解,从而抑制cGAS-STING介导的免疫激活。作者还确立USP7可作为预测放射免疫治疗反应的生物标志物,并证实靶向USP7能够增强放疗联合免疫检查点阻断的疗效。这些发现为放疗-免疫联合疗效差异提供了机制性解释,并提出通过干预USP7-TREX1轴克服免疫治疗耐药的新策略。
参考文献:
Li L, Ye Q, Ma J, Wang Z, Liu T, Lei Y, Lu M, Kang J, Xiang H, Li B, Xu S, Wang K, Chen Y, Chen J, Ma B, Huang W, Cai M, Wu N, Li Y, An J, Jiang C, Ye R, Liu J, Lin SH, Gao Y, Ma J, Li L. Ubiquitination-directed cytosolic DNA degradation governs cGAS-STING-mediated immune response to DNA damage. Cancer Cell. 2026 Jan 8:S1535-6108(25)00547-1. doi: 10.1016/j.ccell.2025.12.013. Epub ahead of print. PMID: 41512867