






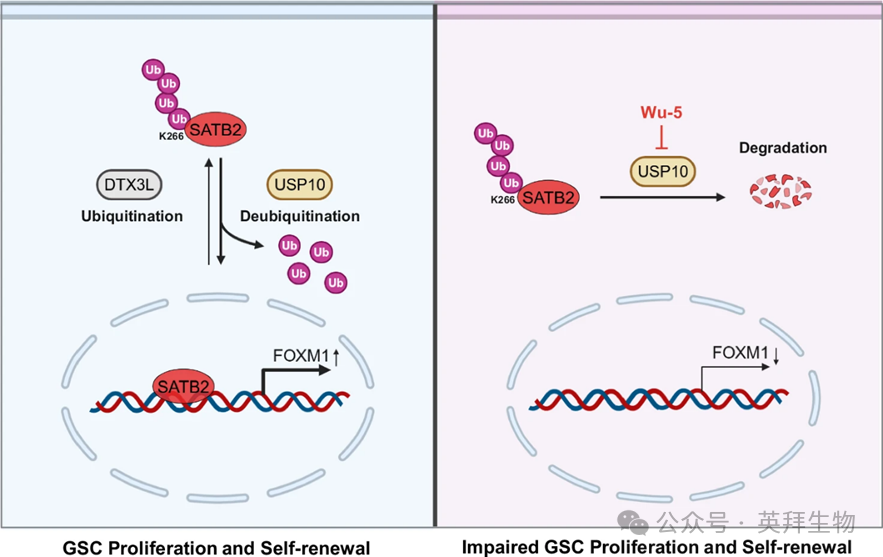
胶质母细胞瘤耐药与复发关键机制曝光:USP10拮抗DTX3L,稳定SATB2
胶质母细胞瘤(GBM)是成人中最常见且最具破坏性的脑肿瘤。GBM的标准治疗是手术,随后进行放疗和化疗。尽管其他实体肿瘤治疗取得了显著进展,但GBM患者的预后依然黯淡,患者的中位生存期仍不足16个月。 GBM的恶性特征被认为与胶质瘤干细胞(GSCs)有关,针对GSCs的新治疗策略开发有望带来重要见解,提升GBM治疗的疗效。
特殊富含AT的序列结合蛋白2(SATB2)是一种核基质相关的转录因子,与DNA的核基质连接区相互作用,从而调控染色质重塑和基因转录。在GBM中,SATB2在GBM组织中高表达,而与低级胶质瘤相比,且主要富集于GSCs中19.SATB2的破坏显著抑制GSC的增殖和自我更新。此外,SATB2在GBM进展中起关键作用,并显著促进了替莫唑胺和放疗耐药的发展。然而,GSCs中SATB2失调的具体机制仍不明确。
泛素-蛋白酶体系统是非溶酶体蛋白酶解细胞内蛋白的主要途径。泛素化是一种标记蛋白质降解的过程,在这一调控机制中起着关键的支持作用。这一过程是可逆的,可以通过一类被称为去泛素酶的多种酶介导。USP10是泛素特异性蛋白酶(USP)家族的成员。最近一项研究表明,USP10的破坏可诱导GBM细胞凋亡,并抑制前神经向间充质的过渡,这是GBM进展中的关键过程。Deltex E3泛素连接酶3L(DTX3L)是一种属于Deltex家族的E3泛素连接酶,与多种癌症的发生和发展有关。然而,USP10和DTX3L在GSC生物学中的功能作用仍然很大程度上是个谜。
该研究2026年1月发表在《Nature Communications》,影响因子:15.7。
技术路线:

主要研究结果:
1、USP10和DTX3L与GSC中的SATB2相互作用
为了确定SATB2蛋白是通过泛素-蛋白酶体途径降解还是溶酶体降解途径被降解,作者首先在使用环己酰胺(CHX)阻断新生蛋白合成后,检测了两种患者来源的GSC中的SATB2蛋白水平。作者发现SATB2蛋白水平以时间依赖性方式下降。相反,在蛋白酶体抑制剂MG-132治疗后,SATB2的蛋白质水平逐渐升高。然而,在用溶酶体抑制剂巴非洛霉素A1处理后,SATB2的蛋白质水平保持不变。这些结果表明SATB2的降解是通过泛素蛋白酶体系统介导的。为识别调控SATB2泛素化和稳定性的潜在去泛素酶和泛素连接酶,作者进行了质谱分析了抗Myc抗体从过度表达Myc标记SATB2的GSC中提取的SATB2相互作用蛋白。值得注意的是,在潜在SATB2结合伴侣的候选中,作者鉴定出了去泛素酶USP10和E3泛素连接酶DTX3L(图1a、b)。
为验证SATB2与USP10或DTX3L之间的相互作用,作者在与Myc标签共表达SATB2、与Flag标签共表达的细胞中进行了共免疫沉淀(CoIP)实验HEK293T。CoIP检测显示,外源性USP10或DTX3L在外源性SATB2拉下复物中易于检测,反之亦然(图1c–f)。这些相互作用通过显示内源性USP10或DTX3L可在GSCs内源性SATB2的免疫沉淀中被检测得到证实(图1g)。为了确定SATB2-USP10或SATB2-DTX3L相互作用所需的具体区域,作者生成了多种截断突变体的SATB2-Myc、USP10-Flag和DTX3L-Flag(图1h–j)。CoIP检测显示,SATB2(氨基酸232–437)的中心结构域与USP10(氨基酸401–798)的C端USP结构域是相互作用的必要条件。此外,作者的结果还表明,SATB2(氨基酸232–437)和DTX3L(氨基酸201–554)的中心结构域对它们的相互作用是必要的(图1m,n)。综合来看,这些数据表明去泛素酶USP10和E3泛素连接酶DTX3L与GSCs中的SATB2相互作用,表明USP10和DTX3L可能调控SATB2的稳定性。
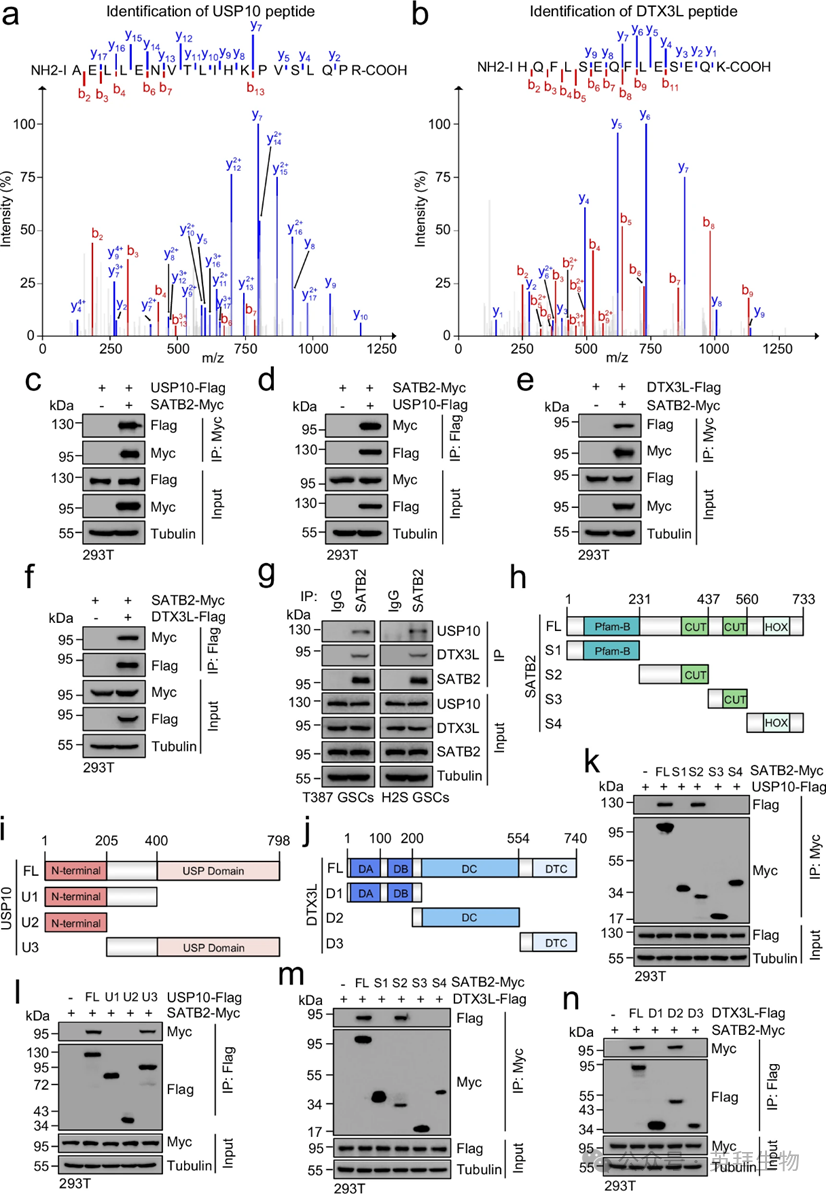
图1:USP10和DTX3L与SATB2相互作用。
2、USP10和DTX3L优先由GSC表示
考虑到SATB2被报道为GSC优先表达。随后,作者旨在验证USP10和DTX3L是否也主要由GBM中的GSC表达。最初在五对匹配的GSCs和非干肿瘤细胞(NSTC)中检测了USP10和DTX3L的蛋白表达。免疫印迹(IB)分析显示,与匹配NSTCs相比,USP10和DTX3L在GSCs中更为突出(图2a)。此外,GSCs中SATB2的富集(图2a),与作者之前的研究结果一致。此外,qPCR分析还显示,GSCs的USP10和DTX3L的mRNA水平表达远高于NSTCs(图2b,c)。接着作者比较了USP10和DTX3L的表达,结果显示USP10在GSC中的表达显著高于DTX3L(图2d)。由于去泛素酶和E3泛素连接酶对蛋白质稳定性具有受阻作用,USP10表达高于DTX3L可能解释了GSCs中SATB2的丰富表达。免疫荧光染色显示,USP10和DTX3L主要位于细胞质中,而SATB2主要存在于GSCs的细胞核中(图2e,f)。这一观察表明,细胞质上的SATB2在去泛素化后可能被转移到细胞核。为验证该假设,作者首先在GSCs中对MG-132或DMSO处理后的SATB2进行了免疫荧光染色。随后,为了确认GSC在GBM中USP10和DTX3L的优先表达,作者考察了多种人类GBM肿瘤中USP10和DTX3L的表达模式。数据确认USP10和DTX3L在表达GSC标记SOX2的胶质瘤细胞中优先表达(图2g、h)。此外,USP10和DTX3L在人类原发性GBM中均与SATB2共定位(图2i、j)。综合来看,这些数据表明,在人类GBM肿瘤中,USP10和DTX3L均由GSC优先表达。
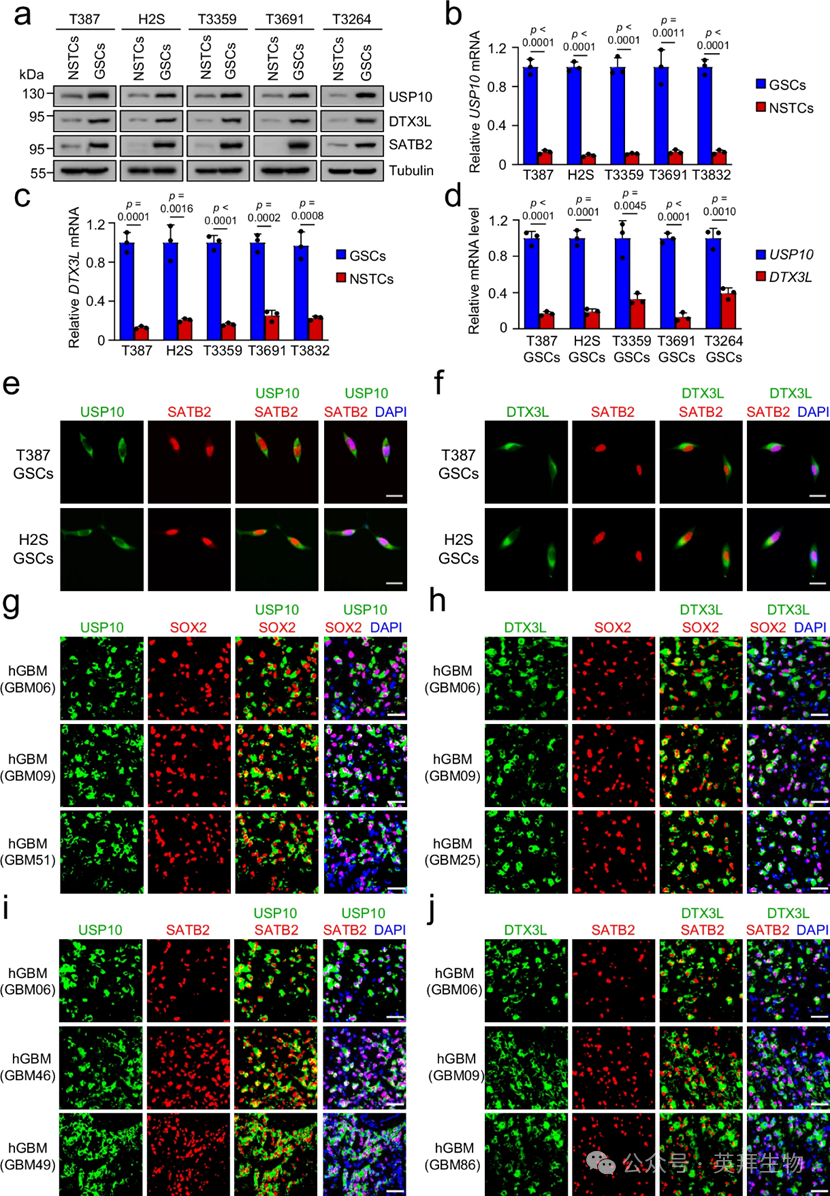
图2:USP10和DTX3L在GBM中由GSC优先表达。
3、USP10是GSC自我更新和GSC驱动肿瘤生长的必要条件
由于USP10在GSCs中富集,作者通过使用两个独立的靶向USP10的shRNA,探讨了USP10在GSC维持和GSC驱动肿瘤生长中的作用。如预期,两种独立的USP10 shRNA的lentiviral介导表达显著降低了GSCs中的USP10蛋白水平,同时也显著降低了SATB2蛋白水平(图3a)。FOXM1是一个重要的下游靶点,介导SATB2促进的GSC增殖和自我更新。由于SATB2对GSC维持至关重要,USP10破坏导致SATB2蛋白表达减少可能影响GSC功能。作者的数据表明,沉默USP10显著抑制了GSC生长(如细胞滴度检测检测),并减少了DNA复制(如EdU合成检测法所示)(图3c)。沉默USP10还通过肿瘤圈形成和体外限制稀释测定法,损害了GSC自我更新(图3d–g)。为了验证USP10的去泛素酶活性是否对GSC功能必要,作者考察了野生型(WT)或催化不活性突变体(C424A, CA)的异位表达是否能挽救USP10破坏带来的影响。作者采用了另一种USP10 shRNA,专门靶向内源性USP10 mRNA(指定为shUSP10-3')的3' UTR,以实现USP10表达的敲低。因此,作者能够同时沉默内源性USP10并过度表达GSCs中的外源性USP10。这些结果表明USP10通过其去泛素酶活性调控SATB2蛋白水平和GSC功能。
为研究USP10对GSC驱动肿瘤生长的体内影响,将表达shNT或shUSP10的GSCs颅内注射至免疫缺陷小鼠的大脑。携带由GSCs衍生的外种移植并表达shUSP10的动物,其生存时间延长且肿瘤体积减少,均优于对照组(图3h,i)。对Ki67和SOX2的免疫荧光染色还表明,破坏USP10显著抑制了GBM异种移植中的细胞增殖并减少GSC数量(图3j–m)。这一发现表明,USP10破坏促进GBM异种移植中GSCs的分化。综合来看,作者的数据表明USP10在促进GSC自我更新和GSC驱动的肿瘤生长中发挥关键作用。
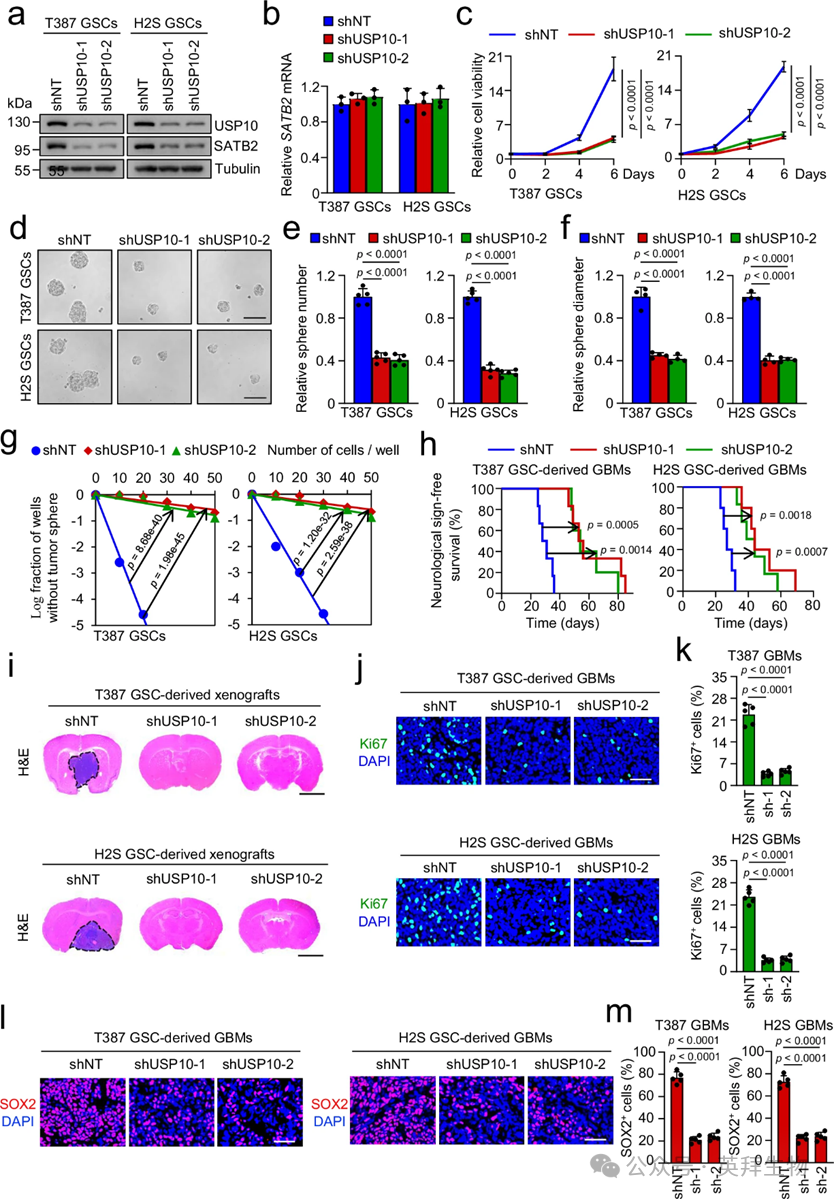
图3:USP10的中断会损害GSC自我更新和GSC驱动的肿瘤生长。
4、DTX3L会损害GSC自我更新和GBM肿瘤生长
由于GSC中DTX3L的表达远低于USP10,作者通过过表达DTX3L来考察DTX3L在GSCs中的作用。作者发现DTX3L的过度表达降低了SATB2蛋白水平,但这种过度表达对SATB2 mRNA水平没有影响(图4a, b)。基于这些发现,作者假设DTX3L也可能影响GSC功能。作者的结果显示,DTX3L的过度表达显著抑制了GSC的生长并减少了DNA复制(图4c–e)。肿瘤球形成和体外限制稀释测定显示,DTX3L的过度表达会损害GSC自我更新(图4f–i)。作者还考察了缺乏E3泛素连接酶活性的DTX3L突变体(C561S和C564S,2CS)是否能调控SATB2蛋白水平和GSC功能。随后作者调查了DTX3L的过度表达是否影响了小鼠大脑中GSC驱动的肿瘤生长。结果显示,携带由DTX3L过度表达GSC产生的肿瘤的小鼠,生存时间延长且肿瘤质量减少(图4j, k)。此外,由DTX3L过度表达的GSC衍生的肿瘤中,增殖细胞和GSC群体数量远少于对照肿瘤(图4l–o)。综合来看,作者得出DTX3L对GSC自我更新和GBM生长具有抑制作用。
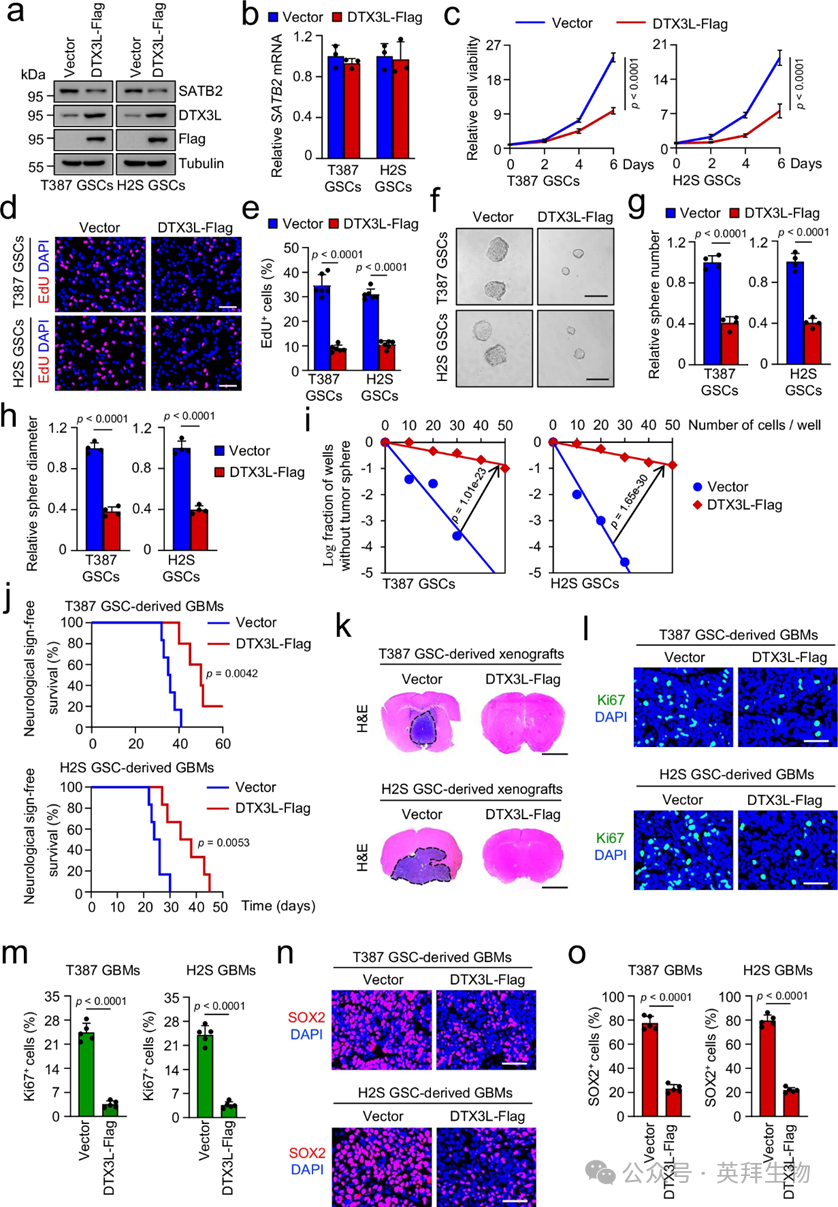
图4:DTX3L的过度表达损害GSC自我更新及GSC驱动的肿瘤生长。
5、DTX3L泛素化SATB2,而USP10介导SATB2去泛素化
作者的结果表明,E3泛素连接酶DTX3L降低了SATB2的蛋白质水平,而去泛素酶USP10负责保持SATB2蛋白水平。为研究DTX3L或USP10对SATB2蛋白稳定性的影响,对DTX3L过表达或USP10沉默GSCs进行了CHX处理。IB分析显示,DTX3L的过度表达或USP10的破坏导致SATB2蛋白的不稳定(图5a-d)。鉴于SATB2通过泛素-蛋白酶体途径降解,作者试图验证DTX3L和USP10通过泛素-蛋白酶体途径调控SATB2的更新。结果显示,MG-132处理抑制了由DTX3L过表达或USP10破坏引起的SATB2蛋白水平下降(图5e,f)。这些数据表明,DTX3L和USP10以蛋白酶体依赖的方式调控SATB2的稳定性。为了确定DTX3L是否确实催化SATB2的多泛素化,作者在HEK293T细胞中共表达了SATB2-Myc和DTX3L-Flag以及His泛素。如图所示。5g,DTX3L的过度表达促进了HEK293T细胞中SATB2的泛素化。一贯地,DTX3L的过度表达促进了GSCs中SATB2的泛素化(图5h)。随后,作者在HEK293T细胞中共同表达了SATB2-Myc和USP10-Flag以及His-泛素。结果表明,USP10的过度表达降低了HEK293T细胞中SATB2的泛素化(图5i)。此外,USP10的破坏增强了GSCs中SATB2的泛素化(图5j)。泛素上的七种不同的Lys残基可实现多泛素化。因此,作者生成了具有完整K6、K11、K27、K29、K33、K48或K63连接的泛素载体,以研究SATB2蛋白上哪种类型的连接被DTX3L或USP10调控。DTX3L强劲地刺激了K48连接多泛素化的组装,而对其他泛素连接类型则无影响(图5k、l)。进一步分析显示,USP10特异性地分解了K48连接的多泛素化,而未影响其他多泛素链类型(图5m,n)。这些结果表明,DTX3L促进SATB2的K48联多泛素化,而USP10则阻止这种多泛素型SATB2。
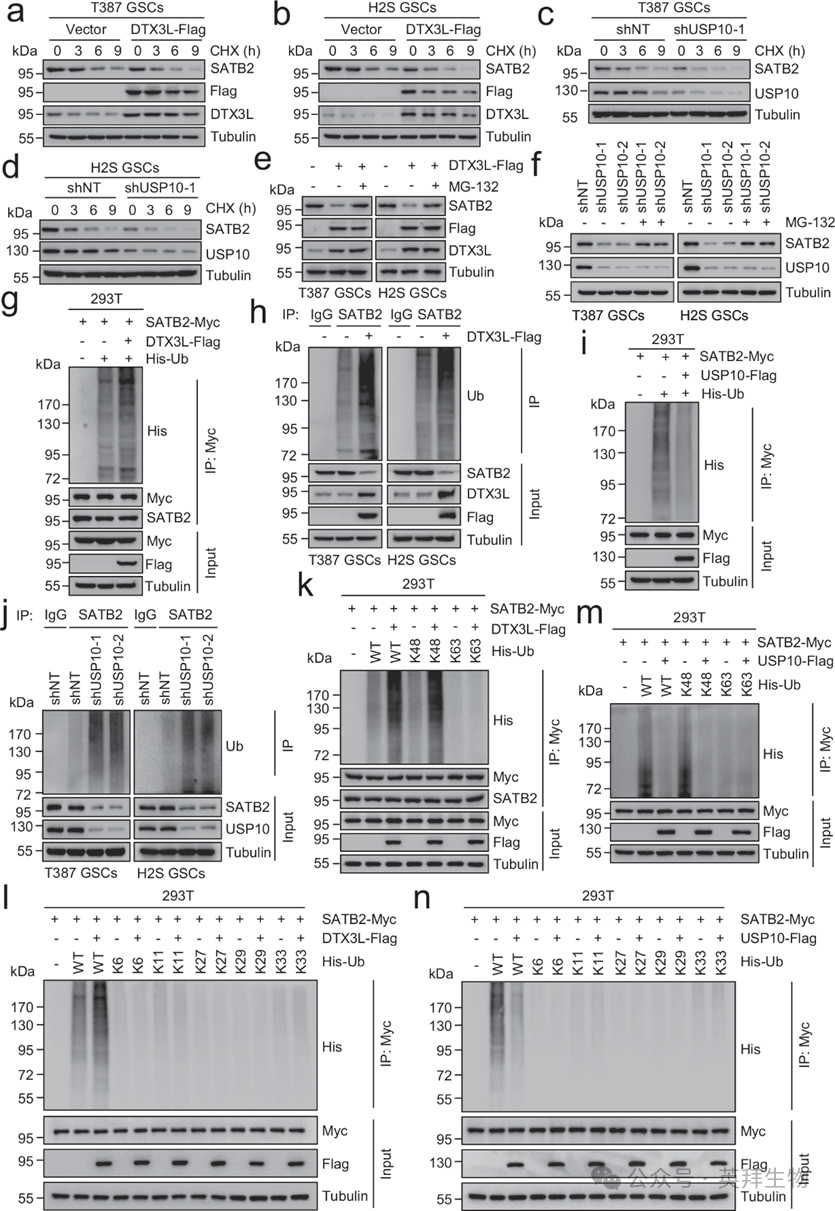
图5:DTX3L介导SATB2泛素化,而USP10通过去泛素稳定SATB2蛋白。
6、SATB2的过度表达可以挽救USP10破坏或DTX3L过表达所带来的影响
鉴于USP10的破坏或DTX3L的过度表达降低了SATB2蛋白水平,作者接着探讨了外源性SATB2的过度表达是否能挽救USP10破坏或DTX3L过度表达引起的效应。通过引入额外的SATB2,通过引入额外的SATB2,将USP10破坏或DTX3L过表达引起的SATB2水平降低,以匹配对照组内源性SATB2水平(图6a、b)。SATB2的外源性过度表达挽救了由USP10破坏或DTX3L过表达引起的GSC增殖和自我更新受损(图6c-h)。重要的是,SATB2的过度表达大大减弱了携带由USP10沉默或DTX3L过度表达GSC衍生异种移植小鼠的生存率提升(图6i、j)。一贯地,SATB2的过度表达能够恢复由USP10破坏或DTX3L过表达受损的GSC驱动肿瘤生长(图6k,l)。此外,免疫荧光染色证实,SATB2的过度表达恢复了因USP10破坏或DTX3L过度表达而受损的体内细胞增殖,并挽救了由USP10破坏或DTX3L过度表达导致的GSC群体减少(图6m–p)。综合来看,这些数据表明,SATB2的过度表达能够挽救USP10破坏或DTX3L过表达引起的影响,表明USP10和DTX3L通过调控SATB2蛋白水平,调节GSC自我更新和GSC驱动的肿瘤生长。
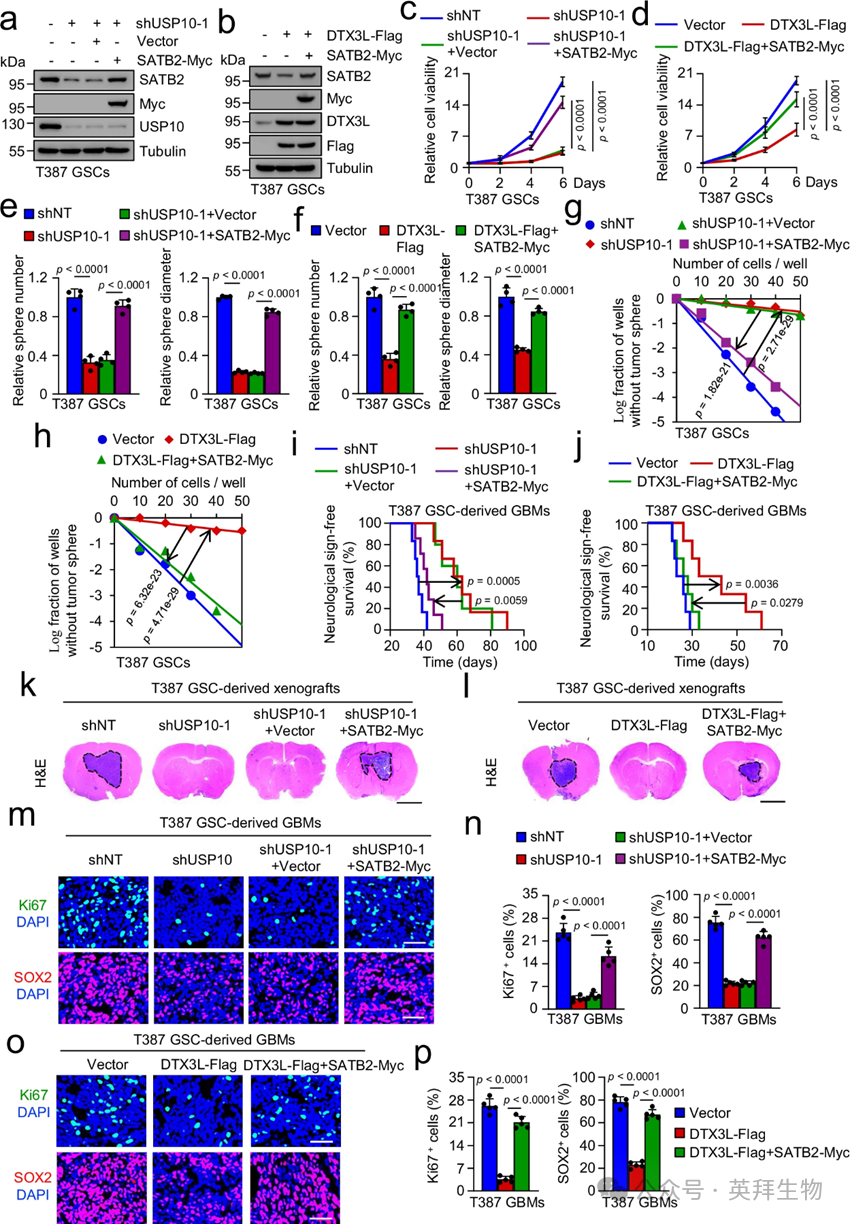
图6:异位表达SATB2挽救了USP10破坏或DTX3L过表达引起的效应。
7、USP10和DTX3L共同调控K266处SATB2的泛素化
为了精确定位人类SATB2蛋白中受USP10或DTX3L调控的赖氨酸残基,作者生成了一系列截断的SATB2突变体并将其导入HEK293T细胞。值得注意的是,在各种突变体中,只有包含氨基酸232至437的SATB2突变体表达对USP10过表达的反应显著增加(图7a)。此外,DTX3L的过度表达还抑制了该SATB2突变体(氨基酸232–437)的表达,但未影响其他突变体的表达(图7b)。随后,作者将SATB2突变体(氨基酸232–437)中的所有赖氨酸残基单独突变为精氨酸。结果显示,SATB2-K266R突变体独特地未受USP10或DTX3L的过度表达影响(图7c,d)。蛋白质序列比对分析显示,SATB2蛋白中含有K266的结构域在七个不同物种中高度保守(图7e)。为确认SATB2的泛素位点,作者进行了质谱分析共表达SATB2-Myc和His-泛素的HEK293T细胞中免疫沉淀的SATB2蛋白。结果显示,泛素基层仅在K266残基上被检测到(图7f),确认K266位点是SATB2的泛素化位点。为了确定 GSCs 中 SATB2 的 K266 是否受 USP10 或 DTX3L 调控,作者在全长 SATB2 中突变了 K266,并将该突变体引入 GSCs。结果证实,GSCs中SATB2-K266R突变体的表达未受USP10破坏或DTX3L过表达影响(图7g–j)。此外,为验证DTX3L是否能直接催化K266残基上的SATB2泛素化,作者在HEK293T细胞中与DTX3L-Flag和His-泛素共表达Myc标记的WT SATB2或K266R(KR)突变SATB2。CoIP实验表明,DTX3L的过度表达促进了SATB2泛素化,而对SATB2-K266R突变体的泛素化则无明显影响(图7k)。此外,USP10的过度表达减少了SATB2泛素化,且对SATB2-K266R突变体的泛素化无影响(图7l)。这些数据共同表明,SATB2中的K266对USP10和DTX3L的去泛素化和泛素化至关重要。
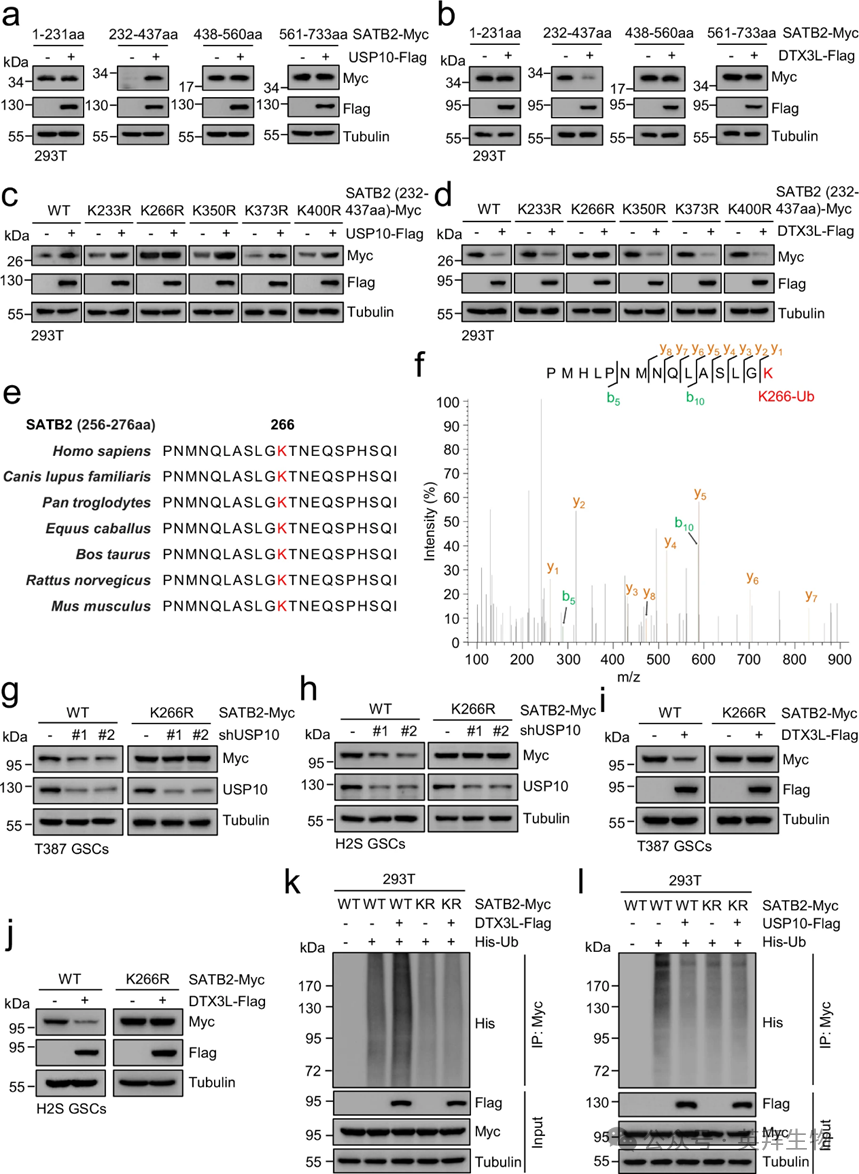
图7:USP10/DTX3L调控K266处SATB2的泛素化。
8、USP10抑制剂Wu-5抑制GSC自我更新及GSC驱动的肿瘤生长
为评估USP10在GBM中的治疗潜力,作者研究了一种名为Wu-5的小分子抑制剂对USP10的药物抑制是否会影响GSCs及GSC驱动的肿瘤生长。细胞存活能力测定显示,8μ米和16μ米的Wu-5剂量依赖性抑制GSC生长,而较低浓度(2和4μ米)则无显著效果(图8a)。Wu-5治疗还以剂量依赖性方式下调GSC自我更新并下调SOX2表达(图8b–e )。此外,Wu-5(8和16μM)剂量依赖性诱导的GSC凋亡,这一点通过凋亡标志物Cripd Caspase-7和Cripd PARP的升高水平得以证明(补充图)。如预期,Wu-5处理以剂量和时间依赖性方式降低了SATB2蛋白水平(图8f)。作者还观察到Wu-5处理以剂量和时间依赖性的方式降低了USP10蛋白水平(图8f),这与以往研究的结果一致 37,38.然而,8μ米和16μ米Wu-5治疗对SATB2和USP10 mRNA水平无影响(图8g)。Wu-5在8和16微M剂量依赖性下,一致地提高了GSCs中SATB2的泛素化(图h)。为确定SATB2过表达是否能挽救Wu-5处理对GSC维持的影响,作者用SATB2-Myc转导GSCs,并用Wu-5(16μM)处理。数据表明,Wu-5处理通过USP10-SATB2轴抑制GSC维持。随后,作者评估了Wu-5对携带GSC衍生异种移植小鼠的治疗效果。与作者的体外研究结果一致,Wu-5治疗有效延长了携带GSC衍生肿瘤的小鼠的存活时间,并减少了小鼠大脑中的肿瘤体积(图8i–k)。Wu-5 施用还显著减少了肿瘤中 Ki67 阳性增殖细胞和 SOX2 阳性 GSC 群体(图8l–0)。为确认Wu-5是否能穿透血脑屏障,作者对BALB/c裸小鼠腹腔内注射Wu-5或DMSO,随后通过LC-MS/MS在小鼠脑组织中检测到Wu-5。结果表明,接受Wu-5治疗2小时的小鼠大脑组织中Wu-5浓度平均达到673 ng/g,而DMSO处理对照组的大脑中Wu-5水平极低,几乎检测不到。这些数据综合表明,药理学抑制USP10通过促进SATB2不稳定显著抑制GSC自我更新和肿瘤生长,表明靶向USP10可能有效改善GBM治疗。
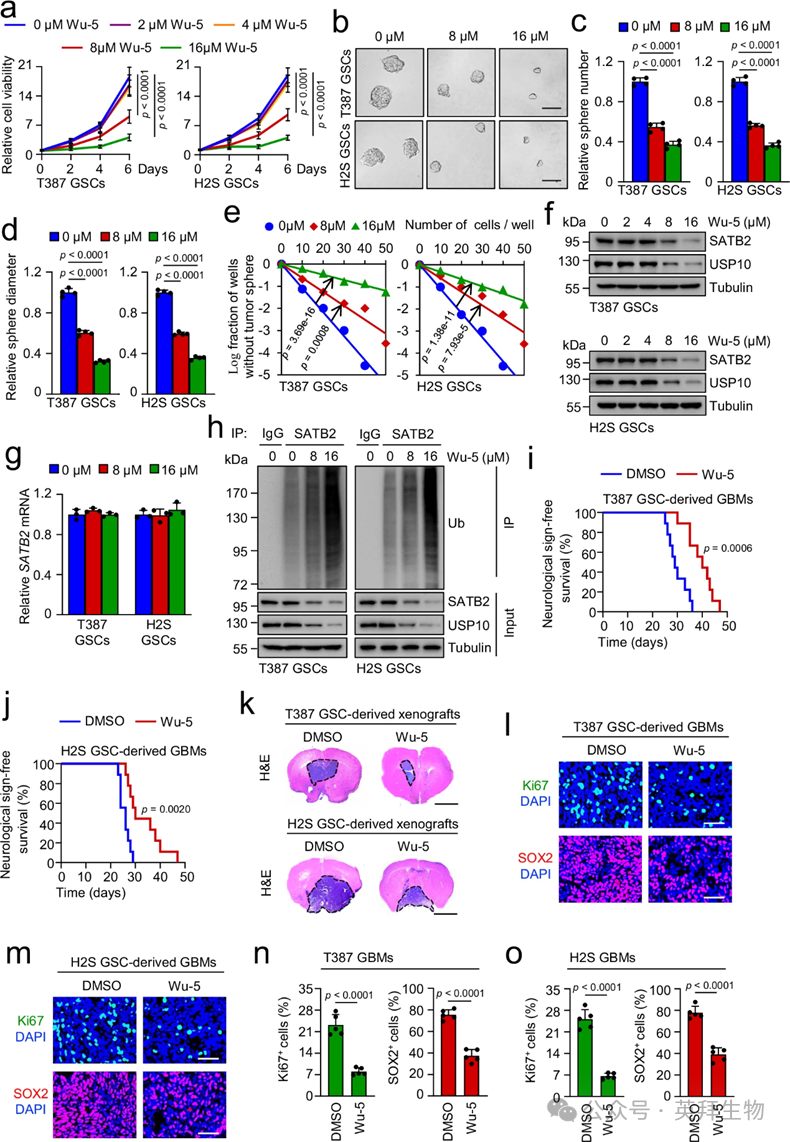
图8:Wu-5治疗抑制GSC自我更新及GSC驱动的肿瘤生长。
结论:
该研究发现去泛素酶USP10与E3泛素连接酶DTX3L协同作用,调控SATB2的泛素化和稳定性,从而调控GSC的维持和肿瘤生长。通过药理抑制USP10来靶向SATB2的稳定性,有望成为提升GBM治疗疗效的有前景的策略。
参考文献:
Guo M, Luo W, Ling P, Lei H, Li L, Duan C, et al. USP10 promotes glioma stem cell maintenance and glioblastoma growth by antagonizing DTX3L-mediated SATB2 ubiquitination. Nat Commun. 2026 Jan 8;17(1):164. doi: 10.1038/s41467-025-67418-9. PMID: 41507172.