






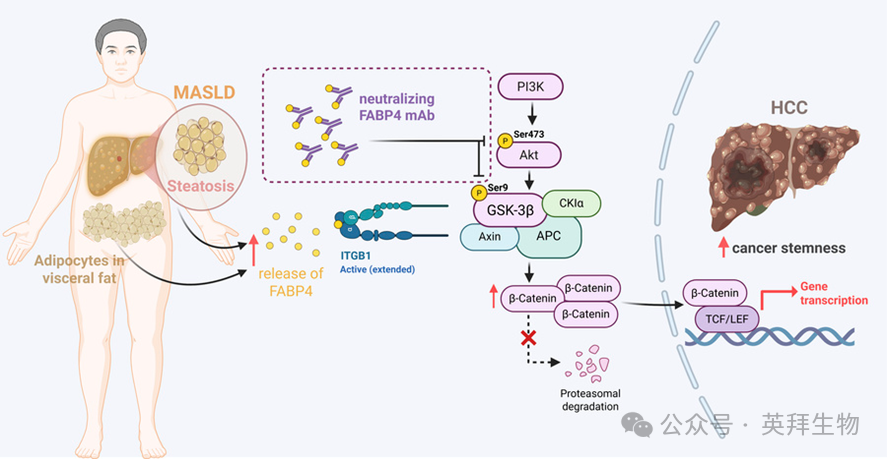
脂肪细胞来源的FABP4通过驱动ITGB1介导的β-catenin激活促进代谢相关脂肪性肝病诱导的肝细胞癌
代谢功能障碍相关脂肪性肝病(MASLD)诱导的肝细胞癌(HCC)是一种与脂肪组织和肝脏脂肪过度堆积相关的新兴恶性肿瘤。理解脂肪细胞在MASLD诱导的HCC发展中的作用至关重要。在一种体外共培养系统中,研究发现分化的脂肪细胞通过旁分泌信号增强了HCC的癌症干细胞特性和耐药性。脂肪酸结合蛋白4(FABP4)主要由脂肪细胞分泌,重组FABP4进一步增强了HCC细胞的癌症干细胞(CSC)特性。值得注意的是,Fabp4-/-小鼠在MASLD-HCC进展中表现出明显的延迟,这与在FABP4表达升高的MASLD患者中观察到的HCC风险增加相关。质谱分析确定了整合素β1(ITGB1)是FABP4的一个结合伴侣。这些数据,连同Fabp4-/-小鼠肿瘤中Wnt/β-catenin通路的显著下调,揭示了FABP4通过ITGB1激活PI3K/AKT/β-catenin信号通路来增强肝脏CSC功能。我们开发了一种抗FABP4中和抗体,它成功地抑制了FABP4驱动的CSC功能并抑制了MASLD诱导的HCC。总之,我们的研究结果表明,脂肪细胞来源的FABP4在MASLD诱导的HCC发展中起着关键作用,靶向ITGB1/PI3K/AKT/β-catenin信号级联反应可能为对抗这种侵袭性疾病提供一种有前景的方法。本文于2025年12月发表于《JOURNAL OF CLINICAL INVESTIGATION》, IF 13.6.
图形摘要
主要实验结果
1.Auranofin直接靶向TOPBP1的BRCT 7-8结构域
通过聚焦于BRCT 7-8结构域,我们旨在寻找TOPBP1的潜在抑制剂。首先,我们采用基于NanoLuc二元技术的蛋白-蛋白相互作用系统(一种结构互补报告系统),筛选了超过2500种FDA批准的药物破坏BRCT 7-8与其同源蛋白配体结合的能力(图1A)。具体而言,将Large BiT(LgBiT)融合到BRCT 7-8,将互补的Small BiT(SmBiT)融合到PHF8的C端片段(PHF8/C),该片段包含一个能够与BRCT 7-8紧密结合的酸性斑块序列(APS)。将这两个独立的模块稳定整合到细胞后,由BRCT 7-8和APS介导的蛋白-蛋白相互作用使分裂的亚基(LgBiT和SmBiT)相互靠近,形成有功能的酶,产生明亮的发光信号。与各种药物孵育4小时后,在活细胞中评估发光情况,初步筛选出47种能将BRCT 7-8与PHF8/C之间相互作用的降低倍数控制在0.4以下的药物(图1B)。使用类似的策略,我们随后用这47种药物进行了第二轮筛选,以测试它们抑制BRCT 7-8与FANCJ C端区域(FANCJ/C)结合的能力,该区域在Thr1133处的磷酸化介导了这种相互作用。这帮助我们将候选药物数量从47种减少到4种(图1C)。已知的TOPBP1抑制剂钙黄绿素-乙酰氧基甲酯(Calcein-AM)被用作阳性对照。
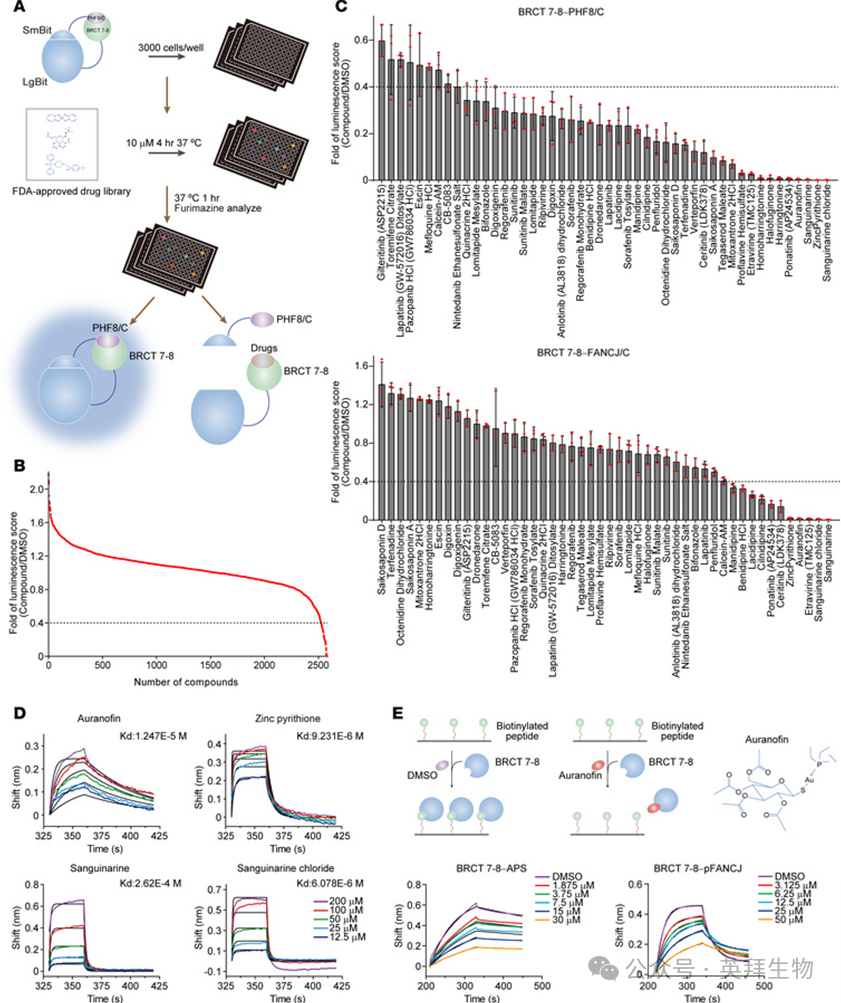
图1 Auranofin直接靶向TOPBP1 BRCT 7-8结构域
为了鉴定能直接靶向BRCT 7-8的小分子,我们接下来采用了生物层干涉技术(BLI),这是一种无需荧光标记即可实时分析生物分子相互作用的光学生物传感技术,用于在体外分析BRCT 7-8与4种候选药物之间的相互作用。结果显示,其中3种药物与从细菌细胞纯化的重组BRCT 7-8结合的解离常数(KD)在近微摩尔范围内(图1D)。为了进一步确定哪一种能在功能上靶向BRCT 7-8,我们通过添加不同浓度的药物,测量了BRCT 7-8与APS肽和磷酸化FANCJ(pFANCJ)肽的亲和力变化。BLI分析显示,与其他两种相比,只有金诺芬——一种含金(I)的磷化合物,能有效破坏BRCT 7-8与测试肽的相互作用(图1E)。这些发现表明,金诺芬有可能占据BRCT 7-8的表面并阻断BRCT 7-8与PHF8和FANCJ两者的相互作用。
2.Auranofin破坏TOPBP1与PHF8和FANCJ的相互作用
为了验证金诺芬在体内的阻断活性,我们首先使用NanoBiT系统测定了金诺芬的抑制效应。稳定表达LgBiT-BRCT7-8和SmBiT-PHF8/C或SmBiT-FANCJ/C的细胞中的发光评分,以剂量依赖的方式被金诺芬降低(图2A)。随后,免疫沉淀结合免疫印迹分析显示,金诺芬显著减弱了FLAG标记的TOPBP1与PHF8和FANCJ的相互作用(图2B)。同样,在较低浓度(2微摩尔)下培养的HeLa细胞中,金诺芬被发现是内源性TOPBP1-PHF8和TOPBP1-FANCJ相互作用的有效阻断剂(图2C)。
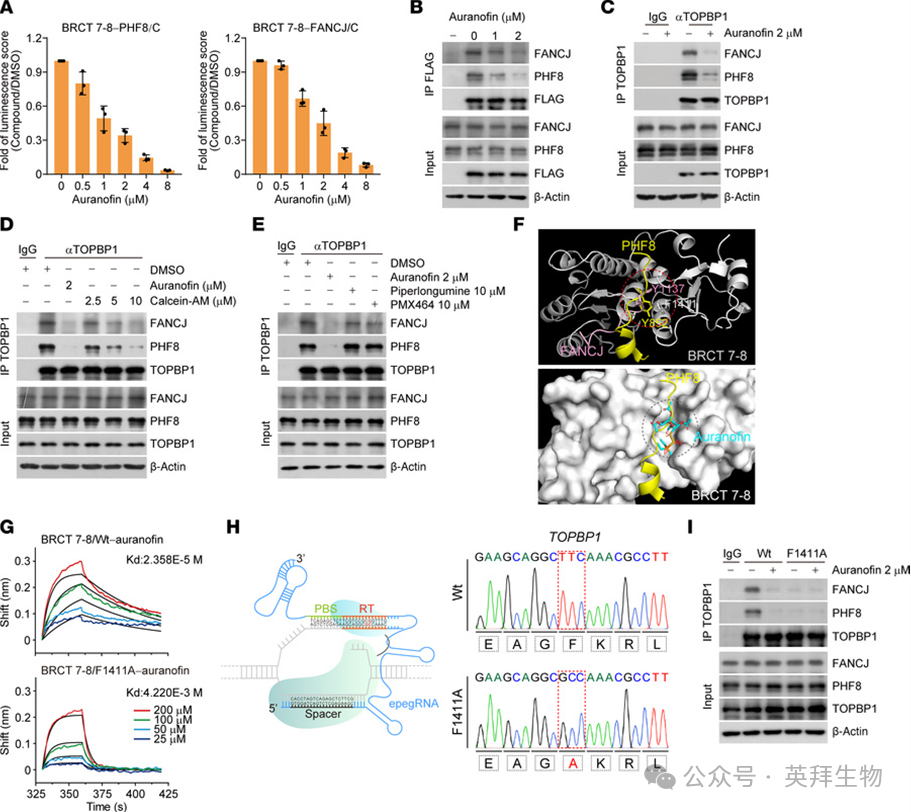
图2 Auranofin破坏TOPBP1与PHF8和FANCJ的相互作用
重要的是,金诺芬对TOPBP1的抑制效果比Calcein-AM强得多(图2D)。此外,我们发现金诺芬的作用独立于其经典靶点硫氧还蛋白还原酶(TrxR),证据是诸如胡椒碱和PMX464等TrxR抑制剂对TOPBP1与PHF8和FANCJ的结合影响微乎其微(图2E)。我们进一步排除了金诺芬对TOPBP1分子相互作用的影响与线粒体通透性相关的可能性。这一结论得到以下观察的支持:作为线粒体通透性阻断剂的环孢素,并不影响金诺芬对TOPBP1与PHF8或FANCJ之间相互作用的抑制作用。
为了进一步理解金诺芬抑制活性的分子机制,我们进行了分子对接分析。与金诺芬的疏水性一致,研究发现它能成功地结合到BRCT 7-8的疏水口袋中(图2F)。由于该口袋中的芳香族残基Phe1411对于PHF8和FANCJ的结合至关重要,我们假设Phe1411应该是金诺芬靶向BRCT 7-8所必需的。为此,我们将Phe1411替换为丙氨酸,并从细菌细胞中纯化了野生型BRCT 7-8(BRCT 7-8/WT)和BRCT 7-8/F1411A。使用这些重组蛋白进行的BLI分析表明,丙氨酸替换(F1411A)显著降低了金诺芬与BRCT 7-8结合的能力(图2G)。为了测试Phe1411在金诺芬结合以及由此引起的蛋白质复合物变形中的重要性,我们采用了Prime编辑工具来修改基因组并生成F1411A编辑的HeLa细胞(图2H)。值得注意的是,金诺芬和F1411A突变能在相似程度上破坏TOPBP1-PHF8和TOPBP1-FANCJ的相互作用,而在F1411A编辑的细胞中,金诺芬未能进一步挑战这些蛋白质复合物的形成(图2I)。由于BRCT 7-8疏水口袋周围没有半胱氨酸残基(图2F),我们认为金诺芬对BRCT 7-8的作用不会基于半胱氨酸中硫醇侧链与金诺芬解离的金离子诱导的共价相互作用。综上所述,这些数据高度表明,金诺芬通过竞争性地结合BRCT 7-8的疏水口袋来破坏TOPBP1与PHF8和FANCJ的相互作用。
我们始终证明,同样依赖于BRCT 7-8的TOPBP1-POLQ复合物的形成,以及有丝分裂期间POLQ向双链断裂位点的募集,都易受金诺芬处理的影响。以下证据支持其分子基础:(a)对共晶体结构与AlphaFold-multimer预测结构的比较表明,与PHF8和FANCJ类似,POLQ中相对保守的芳香族残基F1491与BRCT 7-8疏水口袋中的F1411残基形成π-π堆积相互作用;(b)F1411A突变显著减弱了TOPBP1-POLQ的相互作用;(c)POLQ的F1491残基对其结合TOPBP1和募集到有丝分裂双链断裂位点至关重要。鉴于疏水基序在含有串联BRCT结构域的蛋白质(如TOPBP1、BRCA1和ECT2)的BRCT 7-8界面处具有保守性,我们接下来研究了金诺芬对这些BRCT结构域的特异性。金诺芬对TOPBP1 BRCT 0-2与53BP1、RAD9和HTATSF1之间的相互作用,或TOPBP1 BRCT 4-5与BLM之间的相互作用影响极小甚至没有。此外,我们发现BRCA1和ECT2在金诺芬存在下仍能与FANCJ和RACGAP1有效相互作用。利用生物素标记的金诺芬进行化学蛋白质组学策略,我们鉴定出TOPBP1是首要候选相互作用蛋白之一,而其他含有BRCT结构域的蛋白质则不能被金诺芬捕获。这些发现意味着金诺芬可能特异性靶向TOPBP1的BRCT 7-8区域,这可能涉及到疏水基序之外的特定残基。
3.金诺芬损害TOPBP1募集和ATR激活
考虑到PHF8和FANCJ在复制应激位点处TOPBP1的装载中都扮演重要角色,我们认为金诺芬的施用可能通过解离由BRCT 7-8介导的蛋白质复合物,导致TOPBP1募集缺陷。免疫染色结合共聚焦显微镜分析表明,在金诺芬处理的细胞中,TOPBP1的灶状聚集在喜树碱诱导的复制应激位点处显著受损(图3A)。值得注意的是,这些缺陷无法被ROS清除剂N-乙酰半胱氨酸(NAC)所克服,后者能有效抵消ROS诱导的DNA损伤(图3A)。此外,在喜树碱处理的细胞中,TOPBP1灶状聚集的形成在很大程度上不受TrxR抑制剂胡椒碱和PMX464的影响(图3A)。相比之下,金诺芬对未应激细胞中TOPBP1与活跃复制染色质的结合仅产生轻微改变。这些观察结果表明,金诺芬在功能上抑制了复制应激背景下TOPBP1的装载。
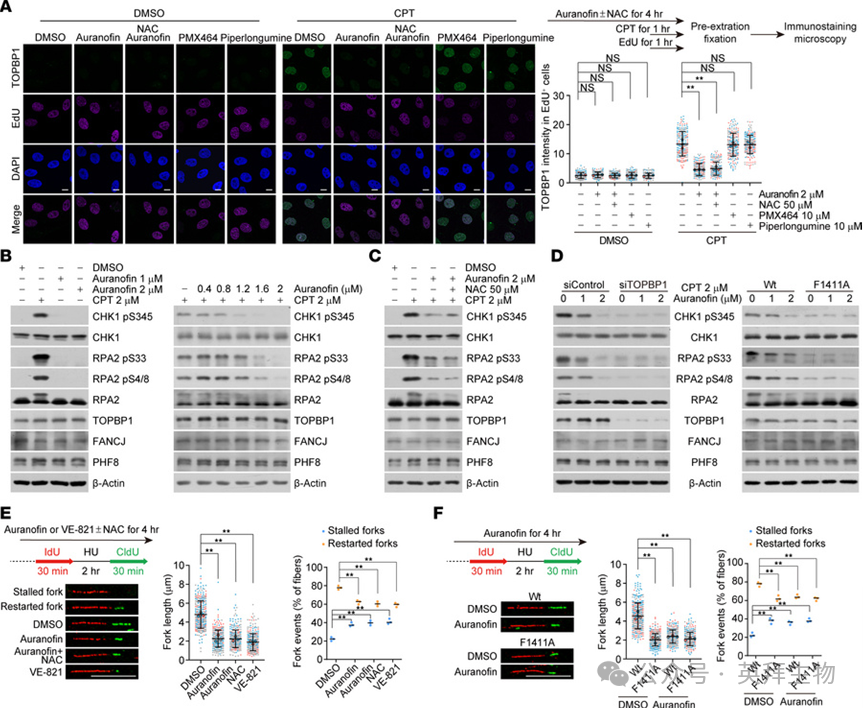
图3 金诺芬损害TOPBP1募集和ATR激活
接下来我们探究金诺芬诱导的TOPBP1装载缺陷是否抑制了ATR活性。首先,我们检测了作为ATR经典底物的CHK1和RPA2的磷酸化水平。免疫印迹分析显示,金诺芬没有刺激而是损害了喜树碱触发的CHK1和RPA2磷酸化,且呈剂量依赖性(图3B),并且这种效应不能被NAC改善(图3C)。重要的是,我们发现在TOPBP1耗竭的U2OS细胞或F1411A编辑的HeLa细胞中,金诺芬未能进一步减弱ATR活性(图3D)。为了进一步评估金诺芬在ATR激活中的抑制作用,我们接下来检测了在缺失或存在金诺芬情况下的复制叉完整性。具体而言,用5-碘-2'-脱氧尿苷(IdU)标记新合成的DNA,然后用核糖核苷酸还原酶抑制剂羟基脲处理,随后在去除羟基脲后用5-氯-2'-脱氧尿苷(CldU)标记。与先前报道及本文显示的ATR抑制结果一致,添加金诺芬导致复制链长度显著缩短、停滞复制叉比例增加以及重启复制叉相应减少(图3E)。同样,这些效应均未被NAC缓解(图3E)。在F1411A编辑的细胞中观察到相似的表型,并且金诺芬处理并未观察到复制完整性的叠加缺陷(图3F)。综上所述,这些数据表明,金诺芬通过破坏TOPBP1的募集来抑制ATR活性。
4.金诺芬解离TOPBP1的液-液相凝聚物
据报道,TOPBP1经历液-液相分离(LLPS),自组装成微米级反应中心以启动ATR/CHK1信号传导。因此,我们想知道金诺芬是否能干扰TOPBP1凝聚物的形成。为此,我们首先纯化了带有GFP标签的BRCT 6-8结构域(包含BRCT 6、ATR激活结构域和BRCT 7-8结构域)。与报道一致,重组BRCT 6-8在体外容易形成液滴,并且它们在相当程度上对金诺芬和1,6-己二醇(1,6-HD,在相分离研究中广泛用作溶解LLPS组装的对照)极其敏感(图4A)。此外,发现F1411A突变显著损害了BRCT 6-8的类液体结构(图4B)。
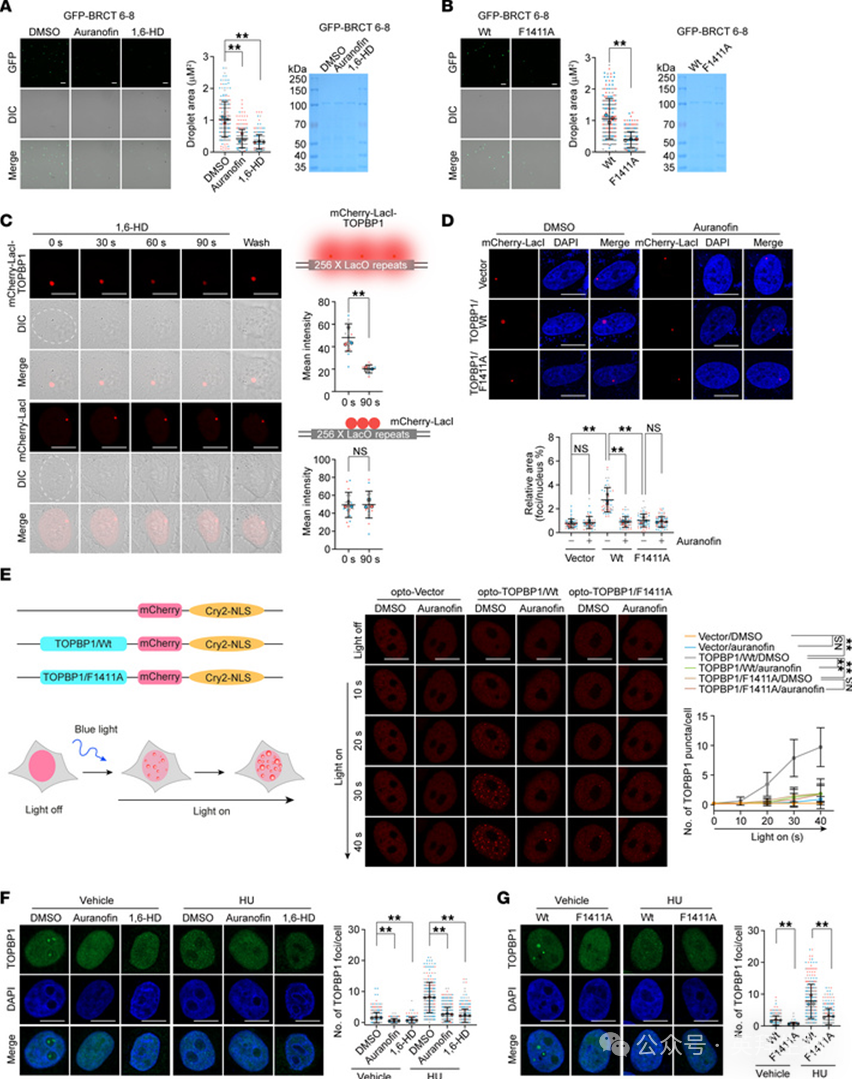
图4 金诺芬解离TOPBP1液-液相凝聚物
随后,我们利用LacO-LacI相互作用系统来测试TOPBP1的相分离。有趣的是,mCherry-LacI-TOPBP1能够形成比mCherry-LacI大得多的斑点(图4C)。与1,6-HD类似,金诺芬处理显著解离了mCherry-LacI-TOPBP1的凝聚物,但对mCherry-LacI没有影响(图4C)。与TOPBP1/WT相比,较小的TOPBP1/F1411A斑块对1,6-HD和金诺芬具有抗性(图4D)。接下来,我们使用一种光遗传学工具评估金诺芬在抑制TOPBP1自组织生物分子凝聚物中的作用。将TOPBP1融合到拟南芥来源的隐花色素2(Cry2),这是一种在488 nm光照射下发生寡聚化的光遗传蛋白(图4E)。结果显示,TOPBP1/WT在活细胞中高效地形成光诱导的光学液滴,而F1411A突变或金诺芬处理几乎完全消除了光遗传学TOPBP1的聚集信号(图4E)。
为了进一步探究金诺芬对TOPBP1生物分子凝聚的影响,我们检测了内源性TOPBP1的LLPS。如报道所示,在生理条件下,TOPBP1在核仁中形成明显的斑点,而这些液滴被金诺芬和1,6-HD破坏(图4F)。在响应羟基脲诱导的复制应激时,TOPBP1表现出许多亚结构化的纳米凝聚物簇,这些簇在很大程度上被金诺芬和1,6-HD瓦解(图4F)。同样,在F1411A编辑的细胞中观察到TOPBP1的相分离缺陷(图4G)。综上所述,这些结果表明金诺芬是TOPBP1液体凝聚的强效抑制剂。
鉴于AAD中的低复杂度区域对TOPBP1凝聚的关键作用,我们提出BRCT 7-8与AAD之间的分子间相互作用,连同较弱的AAD-AAD接触,可能共同促进TOPBP1类液体液滴结构的形成。为了验证这一假设,我们首先分析了AAD的一级序列。我们发现TOPBP1 AAD中的残基Y989和F1071分别对应于PHF8的Y852和FANCJ的Y1137,这两者对于它们与BRCT 7-8的相互作用都至关重要。AlphaFold-multimer分析表明,Y989或F1071都与BRCT 7-8疏水口袋中的F1411形成π-π堆积相互作用。此外,BLI分析证实了这些残基在与BRCT 7-8结合中的必要性,并且金诺芬有效破坏了BRCT 7-8与含有Y989或F1071的肽段的相互作用。与F1071A类似,Y989A突变也削弱了TOPBP1在体外和体内形成凝聚物的能力。值得注意的是,双突变产生的表型与观察到的TOPBP1 F1411A突变或金诺芬处理相似。这些数据表明,BRCT 7-8介导的TOPBP1分子间相互作用对其凝聚物形成至关重要,从而揭示了金诺芬诱导TOPBP1凝聚破坏背后的机制。
我们接着研究了PHF8和FANCJ对TOPBP1 LLPS形成的贡献。首先,我们证明了PHF8和FANCJ在LacO位点周围与mCherry-LacI-TOPBP1共同聚集成显著的焦点。这些焦点对1,6-HD或金诺芬处理敏感。此外,敲低PHF8或FANCJ会导致TOPBP1凝聚物体积的轻微减小。这些观察结果表明PHF8和FANCJ可以整合到TOPBP1凝聚物中并促进其液体凝聚。总的来说,我们提出金诺芬通过同时中断TOPBP1的分子间相互作用以及TOPBP1与其蛋白质伴侣的关联,来抑制TOPBP1凝聚物的组装。
5.金诺芬阻止RPA装载到受干扰的复制叉
鉴于复制应激期间RPA沉积到ssDNA需要TOPBP1-FANCJ相互作用,且该相互作用对金诺芬敏感,我们接下来试图确定金诺芬是否在RPA装载中起不利作用。免疫荧光分析显示,在喜树碱诱导的复制应激位点,RPA2以及RPA2 pS33的灶状形成被金诺芬显著损害,但未被胡椒碱和PMX 464损害,而NAC处理对金诺芬触发的RPA装载缺陷几乎没有影响(图5A)。在检查F1411A编辑细胞中RPA1和TOPBP1的募集时得到了类似结果(图5B)。一致地,在羟基脲处理的细胞中,无论是否存在NAC,金诺芬都导致染色质结合的RPA和TOPBP1大幅减少(图5C)。随后我们证明,生物素标记的金诺芬既不能捕获FANCJ也不能捕获RPA,并且FANCJ-RPA复合物的形成对金诺芬具有抗性。尽管有报道称金诺芬抑制蛋白酶体以及蛋白酶体相关的去泛素化酶UCHL5和USP14,但似乎硼替佐米对蛋白酶体的抑制或UCHL5/USP14的敲低均未明显改变TOPBP1、RPA2和RPA2 pS33的灶状形成,这表明金诺芬在干扰复制应激反应时独立于泛素-蛋白酶体系统起作用。
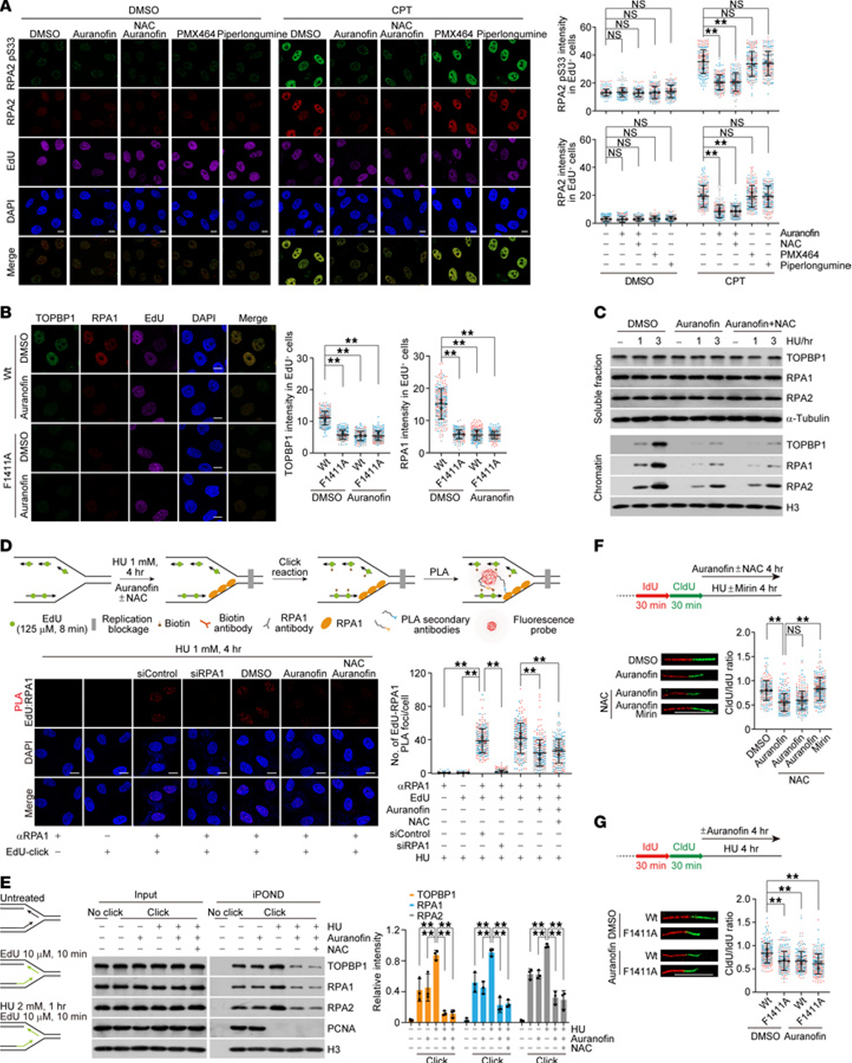
图5 金诺芬阻止RPA装载到受干扰的复制叉
我们接下来在DNA复制叉处进行了蛋白质相互作用的原位分析,以进一步评估金诺芬是否阻断新生复制叉处的RPA装载。简而言之,用羟基脲处理HeLa细胞以诱导复制叉停滞,并使用针对RPA1和一抗及生物素化EdU的邻近连接技术检测RPA与新生ssDNA之间的相互作用,这样只有当RPA位于新生ssDNA 40纳米范围内时才会产生有效的RPA1-EdU邻近连接信号。结果显示,RPA1定位于复制应激位点,而在金诺芬处理下,RPA1-EdU信号显著减少,且不受NAC干扰(图5D)。当通过分离新生DNA上蛋白质的实验检测RPA1、RPA2和TOPBP1在受阻复制染色质处与新生复制叉的关联时,也得到了相同的结果(图5E)。然而,凝胶迁移实验表明,重组RPA与ssDNA的结合不受金诺芬影响,排除了金诺芬直接阻断RPA-ssDNA相互作用的可能性。非变性BrdU染色显示,加入金诺芬后,在复制应激下BrdU灶的形成显著增加。这表明ssDNA积累增多,可能源于ATR失活导致的新复制起点过度激活,或RPA保护丧失引起的ssDNA暴露。因此,金诺芬处理细胞中RPA装载的缺陷不可能是ssDNA生成减少的结果。这些结果表明,金诺芬间接抑制了RPA在复制应激位点向ssDNA的装载,至少部分是通过破坏TOPBP1支架蛋白复合物实现的。
随后进行了DNA纤维分析,以单分子分辨率评估由金诺芬诱导的RPA装载缺陷导致的复制叉去保护。用IdU和CldU依次标记HeLa细胞,然后用羟基脲处理4小时。测量连续标记IdU和CldU的DNA纤维轨迹长度,并通过相邻CldU与IdU的长度比值反映复制叉稳定性。在对照细胞中,羟基脲处理下的比值接近1,表明停滞复制叉的完整性在复制应激期间基本未受影响。相比之下,金诺芬处理细胞的CldU/IdU比值显著降低(图5F),暗示停滞复制叉的保护存在缺陷。添加核酸酶抑制剂米林可防止此现象,但NAC不能(图5F)。与此一致,在F1411A编辑的细胞中观察到停滞复制叉处的新生DNA降解,并且当这些细胞在存在金诺芬的条件下培养时,没有观察到叠加效应(图5G)。这些数据表明,金诺芬通过抑制TOPBP1-FANCJ介导的RPA装载,在复制应激存在时损害了复制叉的完整性。
6.金诺芬增强乳腺癌细胞对化疗药物的敏感性
在我们之前的研究中,我们发现破坏TOPBP1-PHF8相互作用可抑制乳腺肿瘤发生,并使乳腺肿瘤对PARP抑制剂卢卡帕利和铂类药物顺铂产生特异性敏感性。因此,我们研究了金诺芬在乳腺癌细胞中的潜在抗肿瘤作用。首先,用乳腺癌细胞MDA-MB-231提取物进行的免疫共沉淀证实,金诺芬损害了TOPBP1-PHF8和TOPBP1-FANCJ的相互作用(图6A)。接着,免疫印迹分析表明,喜树碱或卢卡帕利诱导的ATR激活被金诺芬剂量依赖性地抑制(图6B),免疫染色及共聚焦显微镜观察显示,金诺芬破坏了MDA-MB-231细胞中RPA向受干扰复制叉的装载(图6C)。一致地,金诺芬显著增加了经喜树碱或卢卡帕利处理细胞的γH2AX水平(图6D)。值得注意的是,我们发现金诺芬对ATR激活、RPA装载和基因组稳定性的抑制作用在很大程度上独立于ROS系统,因为抗氧化剂NAC未能逆转上述任何表型(图6B-D)。
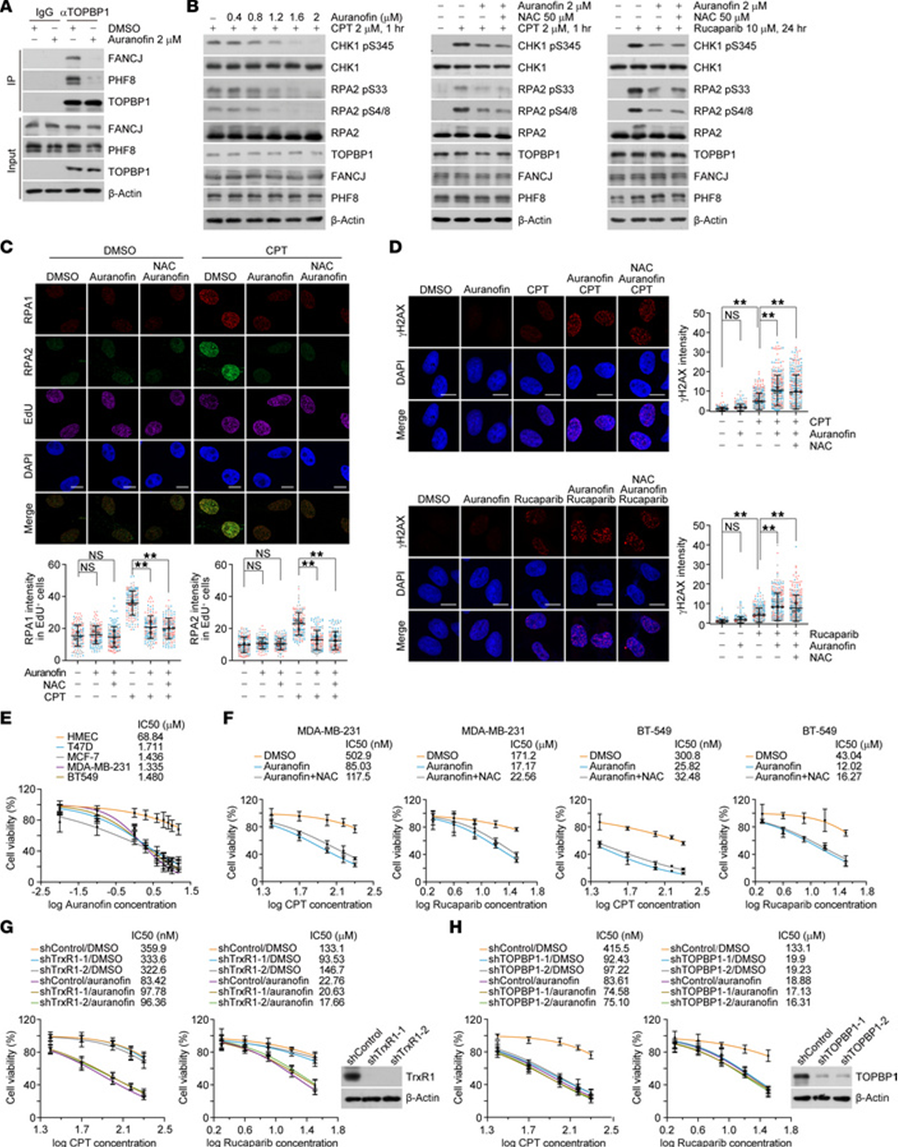
图6 金诺芬增强乳腺癌细胞对化疗药物的敏感性
我们接着检测了金诺芬对细胞存活的影响,并证明多种乳腺癌细胞对金诺芬处理表现出浓度依赖性敏感。相比之下,这种效应在人乳腺上皮细胞(HMECs)中远不明显,突出了金诺芬在乳腺癌细胞中的特异性抗肿瘤作用(图6E)。然后我们研究了金诺芬在乳腺肿瘤化疗中的临床相关性,发现与HMECs不同,金诺芬处理的MDA-MB-231细胞和BT-549细胞对喜树碱和卢卡帕利更敏感(图6F)。重要的是,这种效应不受NAC影响(图6F),且无法通过敲低TrxR来模拟(图6G)。因此,预计金诺芬能将癌细胞的复制应激增加到超过其耐受阈值的水平,从而导致其选择性死亡,同时不影响正常细胞。此外,我们发现金诺芬在TOPBP1敲低的乳腺癌细胞中没有引起更明显的生存缺陷(图6H)。这些发现表明,金诺芬通过直接靶向乳腺癌细胞中的TOPBP1,与喜树碱或卢卡帕利产生协同作用。
7.乳腺肿瘤对金诺芬与卢卡帕利的联合作用具有合成敏感性
为了进一步研究金诺芬的抗肿瘤活性及其与卢卡帕利的合成致死性,我们在NOD/SCID小鼠中建立了MDA-MB-231异种移植瘤,并腹腔注射溶剂对照、金诺芬、卢卡帕利或金诺芬联合卢卡帕利。为了规避金诺芬对TrxR的抑制作用,金诺芬采用低剂量(5 mg/kg)给药。结果显示,单独使用金诺芬或卢卡帕利对异种移植瘤的生长有轻度抑制作用,而两者联用则显著抑制了肿瘤发展(图7A)。接下来,我们将小鼠乳腺癌细胞系E0771注射到C57BL/6J小鼠的乳腺中,以研究金诺芬和卢卡帕利的协同致死性。一致地,与单独使用任一种药物相比,联合给药导致E0771肿瘤显著消退(图7B)。
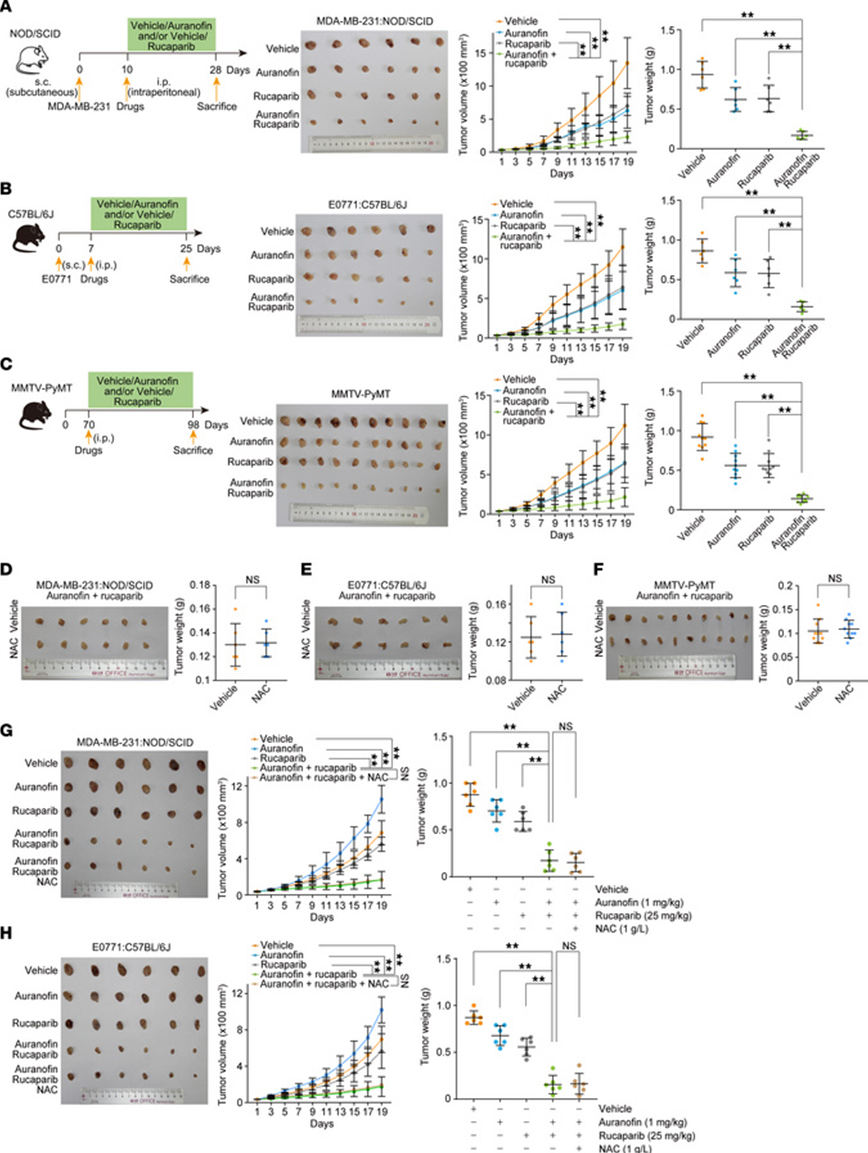
图7 乳腺肿瘤对金诺芬与卢卡帕利的联合作用具有合成敏感性
随后,我们使用了与人乳腺肿瘤具有相似分子和组织学进展的乳腺特异性多瘤病毒中T抗原过表达小鼠模型(MMTV-PyMT)来确认金诺芬的抗肿瘤功能。结果显示,金诺芬显著增强了乳腺肿瘤对卢卡帕利治疗的敏感性(图7C)。ROS诱导的铁死亡似乎不太可能参与此过程,因为低剂量金诺芬给药仅减轻了小鼠的铁过载,而非铁死亡。同时,我们注意到,这三种类型乳腺肿瘤的生长缺陷均不能被NAC给药所改善,进一步提示该协同效应不依赖于引起更高水平的ROS(图7,D-F)。在上述使用不同小鼠肿瘤模型的实验中,各处理组小鼠的体重基本无变化。
此外,金诺芬对E0771肿瘤表现出剂量依赖性效应,且根据小鼠体重、肠上皮结构和增殖以及血液参数的变化,未检测到明显毒性。尽管在金诺芬(5 mg/kg)与卢卡帕利联合治疗的小鼠中观察到了显著的肿瘤消退和极小的毒性,我们仍继续探究临床上相关剂量的金诺芬(按异速缩放法换算,相当于65公斤患者每日6.5毫克,每2天给药1毫克/公斤)与卢卡帕利联用是否能产生协同抗肿瘤效果。结果显示,这种药物联合策略对MDA-MB-231或E0771肿瘤的抑制效果几乎与5 mg/kg金诺芬剂量下观察到的效果相当(图7,G和H)。同时,这些结果对NAC处理表现出耐受性(图7,G和H)。综上所述,这些发现凸显了金诺芬与卢卡帕利联合治疗乳腺肿瘤的转化潜力。
参考文献:
Leung CON, Gurung S, Chung KPS, Leung RWH, Lei MML, Chan MSM, Muliawan GK, Khan SA, Wu XQ, Yu J, Zhu HL, Lu YY, Ma S, Wu X, Hoo RLC, Lee TKW. Adipocyte-derived FABP4 promotes metabolism-associated steatotic liver-induced hepatocellular carcinoma by driving ITGB1-mediated β-catenin activation. J Clin Invest. 2025 Dec 15;135(24):e182322. doi: 10.1172/JCI182322. PMID: 41392988; PMCID: PMC12700556.