






癌症来源的岩藻糖基化外泌体miR-6842-3p通过PTEN/AKT/mTOR/IRF1/CXCL10轴促进ESCC血管生成和转移
肿瘤来源的岩藻糖基化外泌体(FUC-Exo)在癌症进展中发挥重要作用。然而,岩藻糖基化外泌体来源的微小RNA(miRNA)在食管鳞状细胞癌(ESCC)中的功能及机制仍知之甚少。本研究通过小扁豆凝集素(LCA)包被的磁珠分离岩藻糖基化外泌体,结合小RNA测序和RT-qPCR,鉴定出miR-6842-3p是一种新型ESCC生物标志物。结果显示,miR-6842-3p在ESCC组织和血清岩藻糖基化外泌体中表达上调,与临床晚期阶段、较差预后显著相关,可作为ESCC的早期诊断生物标志物。miR-6842-3p具有致癌基因功能,能促进ESCC的肿瘤生长、转移和血管生成。肿瘤来源的岩藻糖基化外泌体miR-6842-3p被HUVECs内化后,会下调PTEN表达,触发AKT和mTOR的磷酸化,进而抑制IRF1表达,最终下调CXCL10表达并驱动血管生成。这些发现表明,miR-6842-3p是ESCC生长、转移和血管生成的关键驱动因子,岩藻糖基化外泌体miR-6842-3p通过介导PTEN/AKT/mTOR/IRF1/CXCL10轴促进血管生成,凸显其作为ESCC新型生物标志物和治疗靶点的潜力。这篇文章于2025年11月发表于《Oncogene》期刊上,IF:7.3。
研究技术路线:
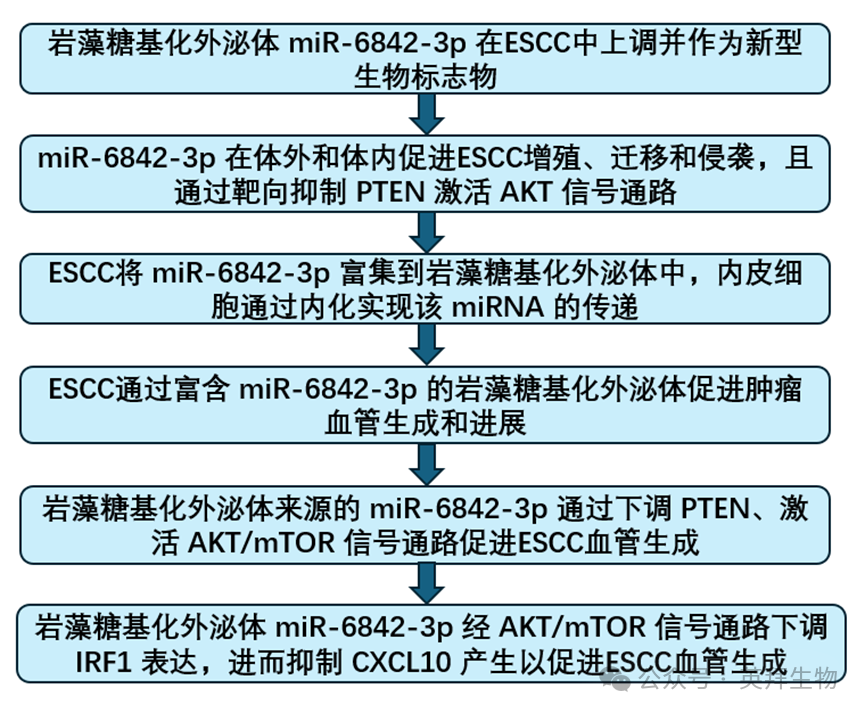
主要实验结果:
1、岩藻糖基化外泌体miR-6842-3p在食管鳞状细胞癌中上调并作为新型生物标志物
为阐明岩藻糖基化外泌体来源的微小RNA(miRNA)在细胞间通讯及食管鳞状细胞癌(ESCC)进展中的作用,作者建立了包含生物标志物发现、筛选、训练、内部验证和外部验证五个阶段的综合方法学框架(图1A)。通过这一严格流程,作者鉴定出四种在ESCC患者血清岩藻糖基化外泌体中差异上调的miRNA(miR-1228-5p、miR-6842-3p、miR-30e-3p和miR-642a-3p),但它们在总外泌体中的表达无显著差异。由于miR-1228-5p、miR-30e-3p和miR-642a-3p在多种恶性肿瘤中已得到广泛研究,而miR-6842-3p的功能作用尚未明确,因此本研究聚焦其在ESCC中的作用。经系统研究和逐步验证,结果表明岩藻糖基化外泌体miR-6842-3p是一种具有潜力的ESCC诊断生物标志物(筛选队列:曲线下面积(AUC)=0.888;训练队列:AUC=0.916;内部验证队列:AUC=0.870;外部验证队列:AUC=0.862),其诊断效能显著优于传统血清生物标志物鳞状细胞癌抗原(SCC-Ag)和癌胚抗原(CEA)(图1B)。尤为重要的是,数据显示早期ESCC患者岩藻糖基化外泌体来源的miR-6842-3p表达水平较健康对照显著升高(图1C、D)。这一独特的表达模式凸显其作为ESCC早期检测可靠指标的潜力(训练队列:AUC=0.908;外部验证队列:AUC=0.875),相较于传统CEA标志物,在早期疾病识别中展现出明显更优的诊断性能(图1E)。此外,分析结果显示,血清岩藻糖基化外泌体miR-6842-3p水平升高与临床晚期阶段显著相关(训练队列:p=0.019,表1;外部验证队列:p=0.037),且与患者较差预后相关(训练队列:p<0.01;风险比(HR)=2.165)(表1,图1F)。同时,与血清表达谱一致,癌组织中miR-6842-3p水平较癌旁正常组织(n=40)显著升高(图1G)。这些综合发现表明,血清岩藻糖基化外泌体miR-6842-3p可能是ESCC早期诊断和预后评估的有价值生物标志物,且可能在ESCC进展中发挥重要作用,值得进一步探究其功能机制。
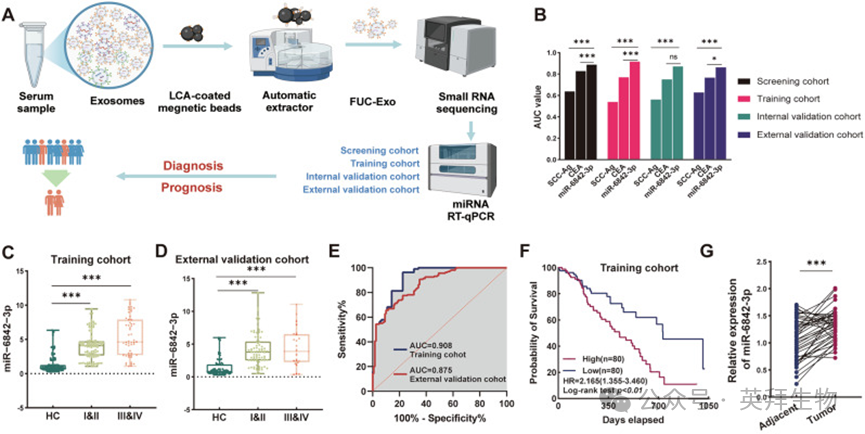
图1. 岩藻糖基化外泌体 miR-6842-3p 在食管鳞状细胞癌中的诊断和预后价值
2、miR-6842-3p在体外和体内促进食管鳞状细胞癌增殖、迁移和侵袭
由于miR-6842-3p在KYSE150细胞中表达较低,在TE-1细胞中表达较高,为进一步阐明miR-6842-3p在ESCC发生发展中的致癌机制,作者分别在KYSE150细胞中转染miR-6842-3p模拟物(mimics),在TE-1细胞中转染miR-6842-3p抑制剂(inhibitor),进行功能获得性和功能缺失性实验。RT-qPCR验证结果显示转染成功。EDU实验、CCK-8实验和集落形成实验结果表明,过表达miR-6842-3p显著增强KYSE150细胞增殖能力,而抑制miR-6842-3p表达则显著减弱TE-1细胞增殖能力(图2A-C)。此外,过表达miR-6842-3p显著提高KYSE150细胞的迁移和侵袭能力,而抑制miR-6842-3p则降低TE-1细胞的这些能力(图2D、E)。作者还检测了增殖相关标志物(PCNA和Cyclin D1)和上皮-间质转化(EMT)相关标志物(N-钙粘蛋白、E-钙粘蛋白和Snail1)。Western blot结果显示,过表达miR-6842-3p可上调ESCC细胞中间质标志物(N-钙粘蛋白和Snail1)和增殖标志物(PCNA和Cyclin D1)的表达,下调上皮标志物(E-钙粘蛋白)的表达,反之亦然(图2F)。这些结果表明,miR-6842-3p通过促进增殖、迁移、侵袭和EMT过程推动ESCC进展。
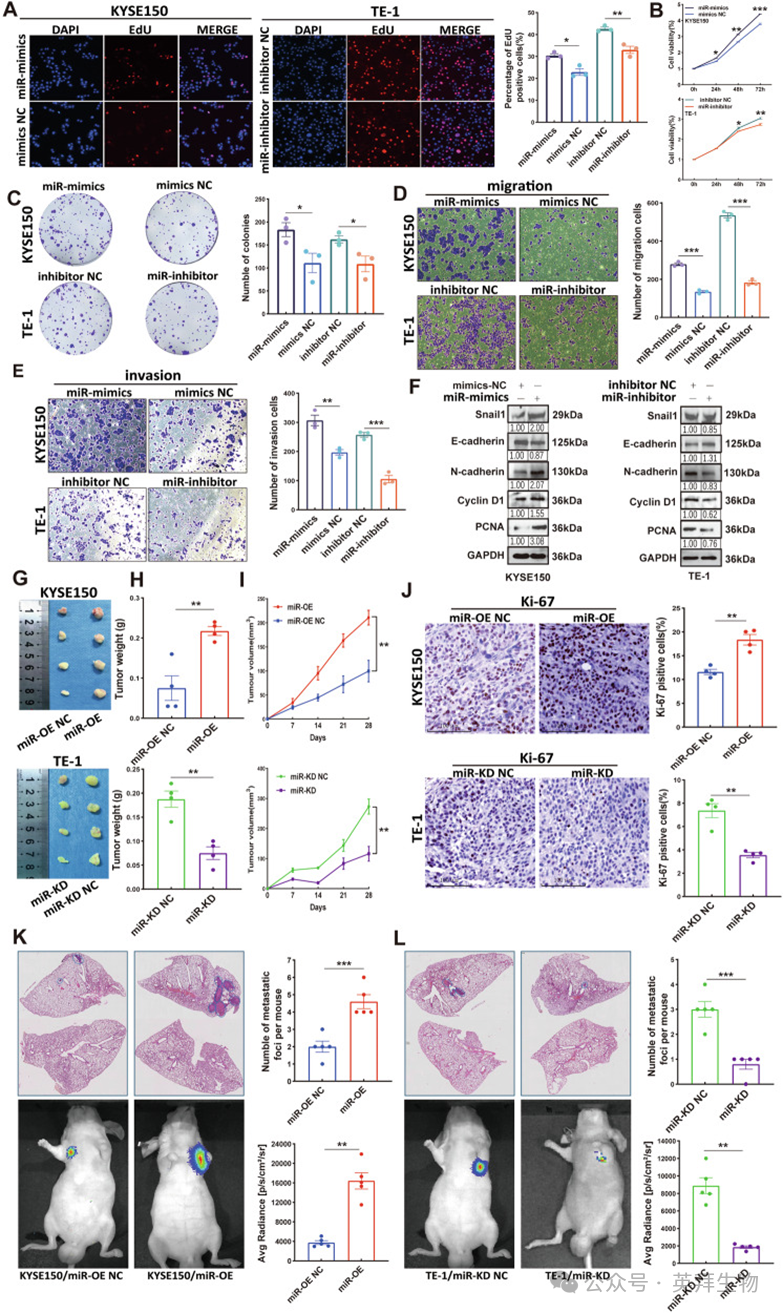
图2.miR-6842-3p在体外和体内促进食管鳞状细胞癌细胞增殖、迁移、侵袭及上皮-间质转化
为验证体外实验结果,作者进行了体内实验。构建稳定转染Lv-miR-6842-3p模拟物(miR-OE)的KYSE150细胞和转染Lv-miR-6842-3p抑制剂(miR-KD)的TE-1细胞,RT-qPCR验证转染效率(。在异种移植模型中,与miR-OE阴性对照(NC)组相比,miR-OE组肿瘤体积和重量显著增大;而与对照组相比,miR-KD组肿瘤体积和重量显著减小(图2G-I)。免疫组织化学(IHC)分析显示,miR-OE组肿瘤组织中Ki-67表达升高,而miR-KD组肿瘤组织中Ki-67表达降低,证实miR-6842-3p具有促肿瘤生长作用(图2J)。在肺转移模型中,过表达miR-6842-3p加速转移进程(肺转移灶增多),而抑制其表达则抑制转移扩散(图2K、L)。综上,这些结果表明miR-6842-3p促进ESCC生长和转移,在ESCC进展中发挥关键作用。
3、miR-6842-3p靶向抑制PTEN表达,激活AKT信号通路,促进食管鳞状细胞癌增殖和转移
为进一步阐明miR-6842-3p调控ESCC进展的机制,作者利用五个mRNA靶标预测数据库(miR TarBase、miRWalk、miRDB、RNA22和RNAInter)筛选miR-6842-3p的潜在下游靶基因。分析鉴定出13个候选靶基因(图3A),其中7个为已知的肿瘤抑制基因。RT-qPCR结果显示,在过表达miR-6842-3p的KYSE150细胞中,这7个肿瘤抑制基因均显著下调,其中PTEN的抑制作用最为显著(图3B)。PTEN作为一种公认的肿瘤抑制基因,在多种癌症中发挥关键作用。
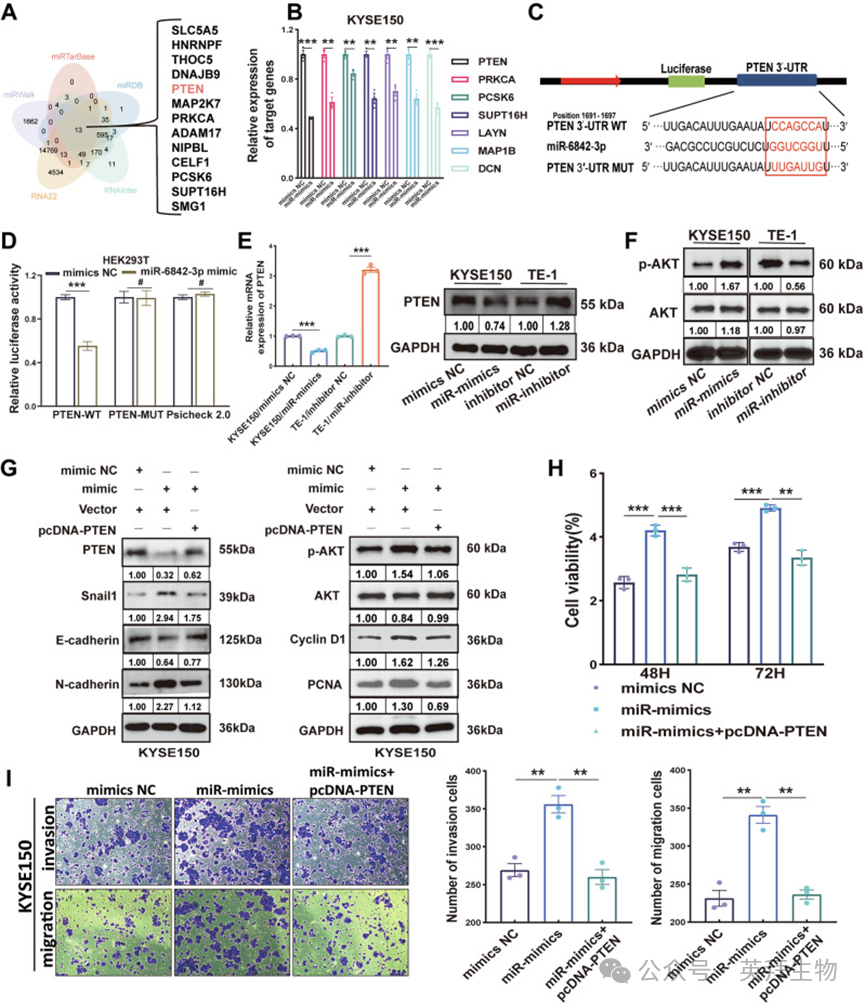
图3.miR-6842-3p靶向抑制 PTEN 表达,激活 AKT 信号通路,促进食管鳞状细胞癌增殖和转移
为证实PTEN是miR-6842-3p的直接靶基因,作者在HEK293T细胞中进行双荧光素酶报告基因实验。结果显示,miR-6842-3p显著降低野生型PTEN 3'非翻译区(3'UTR)的荧光素酶活性,但对突变型PTEN 3'UTR的荧光素酶活性影响极小(图3C、D)。RT-qPCR和Western blot进一步验证显示,过表达miR-6842-3p显著降低KYSE150细胞中PTEN的表达,而敲低miR-6842-3p则升高TE-1细胞中PTEN的表达(图3E)。
AKT通路位于PTEN下游,已有研究表明在人类癌症中PTEN失活常导致AKT激活。Western blot结果显示,过表达miR-6842-3p可下调KYSE150细胞中PTEN表达,上调磷酸化AKT(p-AKT)水平;相反,敲低miR-6842-3p显著升高TE-1细胞中PTEN表达,降低p-AKT水平(图3E、F)。
为明确miR-6842-3p是否通过靶向PTEN激活AKT信号通路,作者进行了挽救实验。Western blot结果显示,过表达PTEN可逆转miR-6842-3p模拟物介导的p-AKT、EMT相关标志物(N-钙粘蛋白、E-钙粘蛋白和Snail1)以及增殖相关标志物(PCNA和Cyclin D1)表达的改变(图3G)。一致地,CCK-8实验显示,过表达PTEN可消除miR-6842-3p对KYSE150细胞的促增殖作用(图3H)。Transwell实验表明,在miR-6842-3p模拟物+PTEN共表达组中,miR-6842-3p模拟物增强的KYSE150细胞迁移和侵袭能力显著减弱(图3I)。综上,这些结果表明miR-6842-3p靶向抑制PTEN表达,激活AKT信号通路,进而促进ESCC增殖和转移。
4、食管鳞状细胞癌细胞将miR-6842-3p富集到岩藻糖基化外泌体中,内皮细胞通过内化肿瘤来源的岩藻糖基化外泌体实现miR-6842-3p的传递
鉴于血管生成在癌症转移中的关键作用,以及越来越多的证据表明外泌体miRNA与多种癌症的转移过程相关,作者进一步探究miR-6842-3p是否通过岩藻糖基化外泌体介导的传递在肿瘤血管生成中发挥作用。首先,RT-qPCR检测不同ESCC细胞中miR-6842-3p水平,结果显示与人脐静脉内皮细胞(HUVECs)相比,miR-6842-3p在ESCC细胞(包括KYSE150、KYSE140和TE-1细胞)中高表达。选择miR-6842-3p表达相对较高的KYSE150和TE-1细胞进行后续实验。通过透射电子显微镜(TEM)、纳米颗粒追踪分析(NTA)和Western blot鉴定从这些细胞条件培养基(CM)中分离的岩藻糖基化外泌体(图4A、B)。为探究ESCC细胞如何在分子水平调控HUVECs,作者首先构建miR-6842-3p稳定过表达的KYSE150和TE-1细胞系。RT-qPCR结果显示,ESCC细胞中过表达miR-6842-3p可导致细胞内及癌细胞来源的岩藻糖基化外泌体中miR-6842-3p水平均升高(图4C、D),表明ESCC细胞将miR-6842-3p富集到肿瘤来源的岩藻糖基化外泌体中。此外,用来自过表达miR-6842-3p的ESCC细胞的岩藻糖基化外泌体处理HUVECs后,HUVECs中miR-6842-3p水平呈时间依赖性升高,提示HUVECs通过内化岩藻糖基化外泌体实现miR-6842-3p的传递(图4E)。外泌体摄取实验进一步证实ESCC细胞来源的岩藻糖基化外泌体可被HUVECs内化,在转染FAM标记的miR-6842-3p过表达载体的KYSE150或TE-1细胞来源的、经WisTracker标记的岩藻糖基化外泌体中,HUVECs内可观察到荧光信号(图4F)。这些结果表明,miR-6842-3p被ESCC细胞包装进岩藻糖基化外泌体,并通过岩藻糖基化外泌体途径传递至HUVECs。
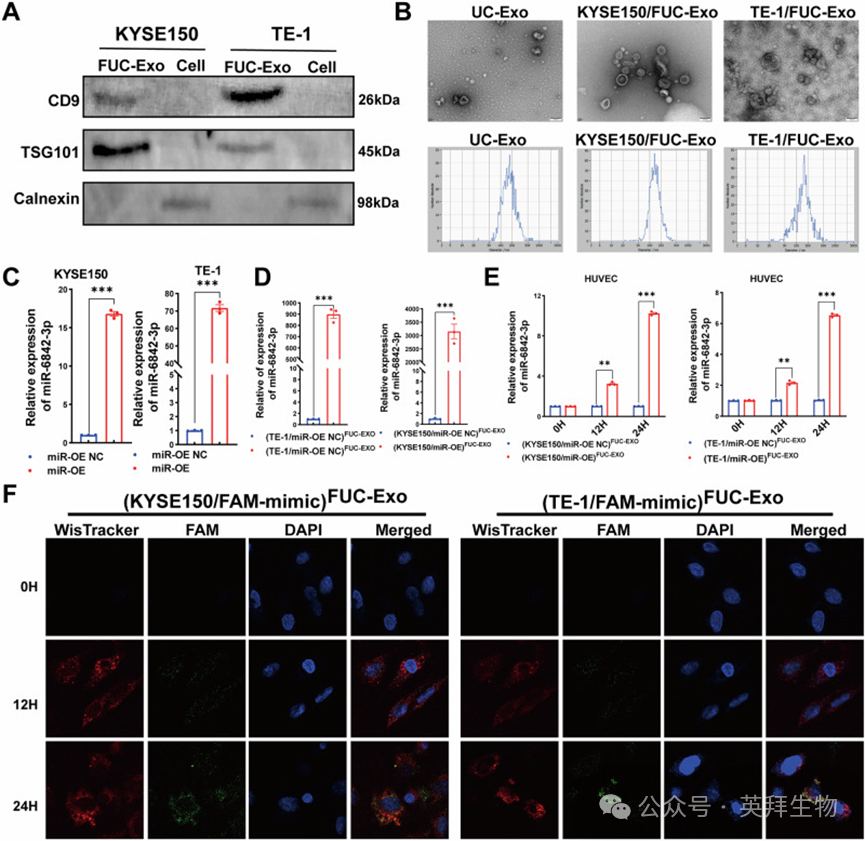
图4.miR-6842-3p通过肿瘤来源的岩藻糖基化外泌体传递至人脐静脉内皮细胞
5、食管鳞状细胞癌细胞通过富含miR-6842-3p的岩藻糖基化外泌体促进肿瘤血管生成和进展
为进一步探究岩藻糖基化外泌体miR-6842-3p在促进肿瘤血管生成中的作用,作者首先向HUVECs瞬时转染miR-6842-3p模拟物或抑制剂以调控其表达,并通过RT-qPCR验证转染效果。管腔形成实验明确显示,miR-6842-3p模拟物增强HUVECs的毛细血管样管腔形成能力,而miR-6842-3p抑制剂则抑制这些细胞的管样结构形成。此外,瞬时转染miR-6842-3p模拟物可上调HUVECs中增殖和血管生成标志物(MMP2、CD31、PCNA和Cyclin D1)的蛋白表达,同时下调紧密连接蛋白(Occludin、ZO-1和Claudin5)的表达,反之亦然。为进一步验证这一观点,作者构建稳定过表达或敲低miR-6842-3p的HUVECs,并通过RT-qPCR检测miR-6842-3p表达。随后进行体内Matrigel栓血管生成实验,将Matrigel皮下注射到小鼠体内。结果显示,过表达miR-6842-3p显著促进Matrigel栓内的新生血管形成;相反,敲低miR-6842-3p则减少迁移至Matrigel栓内的血管数量。作者还检测了异种移植模型肿瘤组织中血管标志物CD31的表达。IHC分析显示,与相应对照组相比,miR过表达(miR-OE)组肿瘤组织中CD31表达显著上调,miR敲低(miR-KD)组肿瘤组织中CD31表达显著下调。这些结果表明,miR-6842-3p水平升高可促进血管生成。
为明确miR-6842-3p是否通过岩藻糖基化外泌体(FUC-Exo)传递发挥其促血管生成作用,作者用外泌体释放抑制剂GW4869预处理KYSE150(miR-OE)细胞。随后从三组细胞中分离岩藻糖基化外泌体:(miR-OE NC)岩藻糖基化外泌体、(miR-OE)岩藻糖基化外泌体和(miR-OE + GW4869)岩藻糖基化外泌体,并用这些外泌体处理HUVECs 24小时。RT-qPCR结果显示,用(miR-OE)岩藻糖基化外泌体处理后,HUVECs中miR-6842-3p水平显著升高,而用(miR-OE + GW4869)岩藻糖基化外泌体预处理后,这种上调作用被消除。这些结果还表明,ESCC细胞通过岩藻糖基化外泌体将miR-6842-3p传递至内皮细胞,导致受体HUVECs中miR-6842-3p富集(图5A)。功能实验(EDU、迁移、伤口愈合实验)显示,(miR-OE)岩藻糖基化外泌体处理显著增强HUVECs增殖和迁移能力,而(miR-OE + GW4869)岩藻糖基化外泌体预处理则显著减弱这些作用(图5B-D)。免疫荧光实验显示,(miR-OE)岩藻糖基化外泌体损害HUVECs中紧密连接蛋白的表达,而GW4869可逆转这一效应(图5E)。此外,(miR-OE)岩藻糖基化外泌体显著促进体外血管生成,而(miR-OE + GW4869)岩藻糖基化外泌体预处理则阻断这一过程(图5F)。这些结果表明,ESCC细胞通过岩藻糖基化外泌体诱导内皮细胞中miR-6842-3p上调,进而增强血管通透性并促进血管生成。
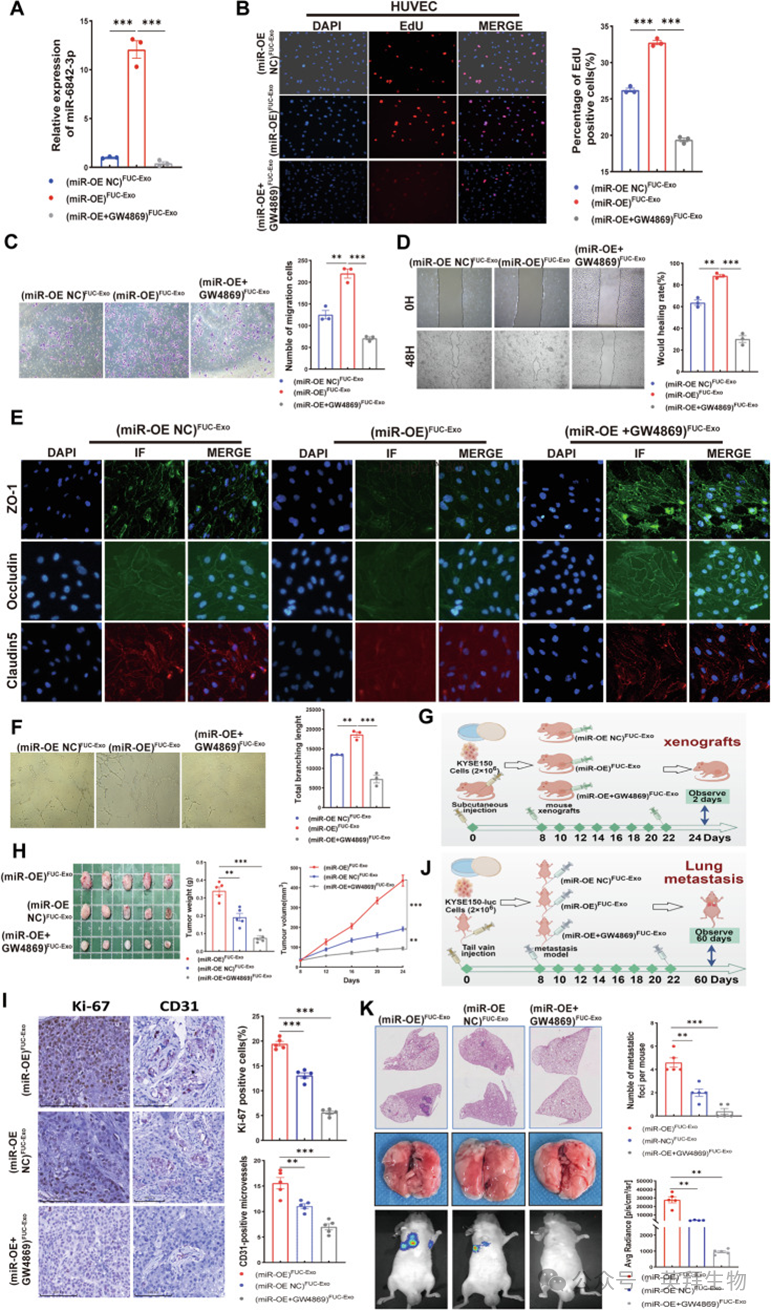
图5.岩藻糖基化外泌体 miR-6842-3p 促进人脐静脉内皮细胞生长、血管生成及食管鳞状细胞癌进展
为验证岩藻糖基化外泌体miR-6842-3p的体内作用,作者建立了肿瘤异种移植和肺转移模型。在异种移植模型中,对携带KYSE150细胞来源肿瘤的小鼠每隔一天给予指定的岩藻糖基化外泌体(5μg)处理(图5G)。对肿瘤大小/体积的评估,以及增殖标志物Ki-67和CD31阳性血管形成的免疫组织化学分析表明,(miR-OE)岩藻糖基化外泌体促进ESCC生长和血管生成。值得注意的是,GW4869有效消除了(miR-OE)岩藻糖基化外泌体的这些促生长和促血管生成作用(图5H、I),表明岩藻糖基化外泌体通过递送miR-6842-3p促进ESCC致癌作用和血管生成。为评估岩藻糖基化外泌体miR-6842-3p对ESCC转移的影响,作者通过尾静脉向BALB/c裸鼠注射2×10^6个KYSE150-luc细胞建立肺转移模型。注射后8天,小鼠每隔一天接受(miR-OE NC)岩藻糖基化外泌体、(miR-OE)岩藻糖基化外泌体或(miR-OE + GW4869)岩藻糖基化外泌体(5μg)处理。两个月后,处死小鼠并分析肺组织(图5J)。转移灶定量结果显示,(miR-OE)岩藻糖基化外泌体促进ESCC转移,而GW4869有效抵消这种促转移作用(图5K)。综上,这些结果表明,岩藻糖基化外泌体介导miR-6842-3p从ESCC细胞向受体细胞的传递,从而促进肿瘤生长、血管生成和转移。
6、岩藻糖基化外泌体来源的miR-6842-3p通过下调PTEN促进食管鳞状细胞癌进展和血管生成
为探究岩藻糖基化外泌体来源的miR-6842-3p是否也通过调控内皮细胞中的PTEN对血管生成产生类似影响,作者首先通过双荧光素酶报告基因实验验证miR-6842-3p与PTEN在HUVECs中的直接调控关系,然后从KYSE150(miR-OE)细胞和TE-1(miR-KD)细胞的条件培养基中纯化岩藻糖基化外泌体。与作者的假设一致,岩藻糖基化外泌体来源的miR-6842-3p可调节HUVECs中PTEN mRNA的表达(。此外,过表达PTEN可减弱岩藻糖基化外泌体来源的miR-6842-3p介导的HUVECs中PTEN mRNA、增殖标志物(PCNA和Cyclin D1)、血管生成标志物(MMP2和CD31)以及紧密连接蛋白(Occludin、ZO-1和Claudin5)表达的改变(图6A、E、F)。与图5中的数据一致,过表达PTEN也可消除岩藻糖基化外泌体来源的miR-6842-3p增强的HUVECs增殖、迁移和管腔形成能力(图6B-D)。这些结果表明,岩藻糖基化外泌体来源的miR-6842-3p通过沉默HUVECs中的PTEN促进血管生成。
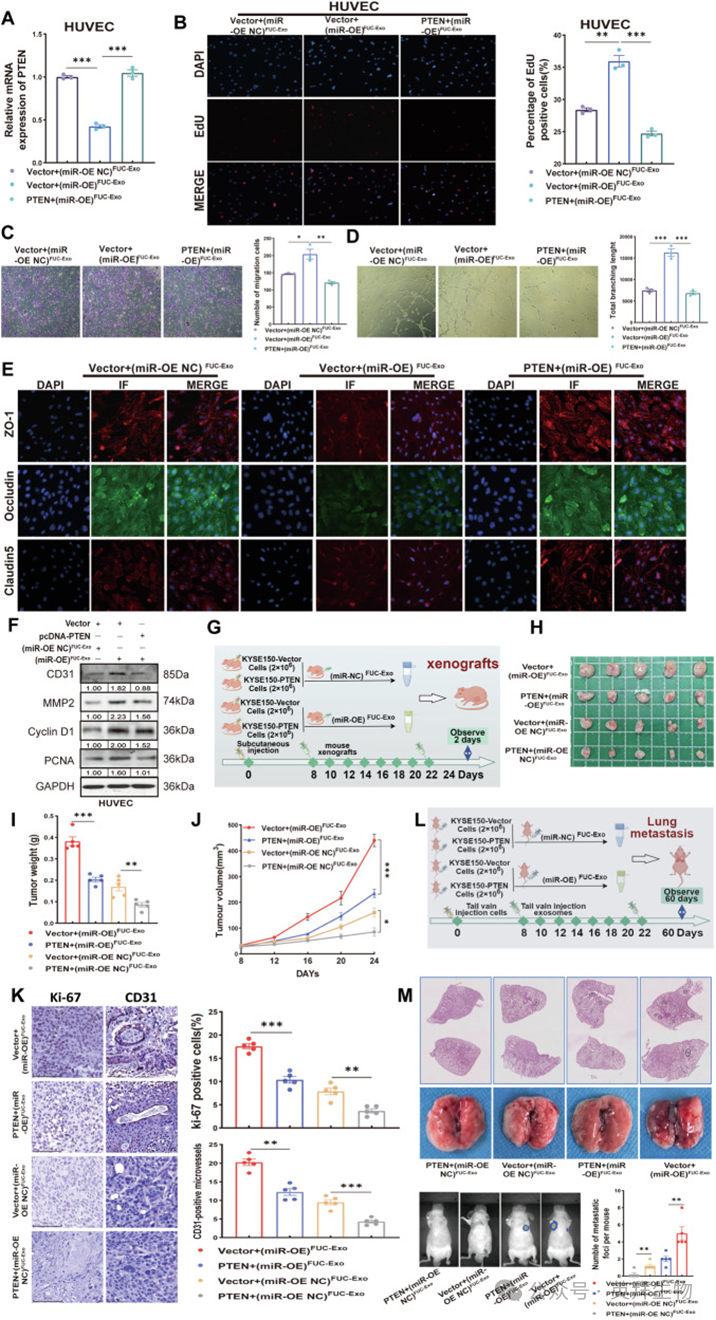
图6.岩藻糖基化外泌体来源的 miR-6842-3p 通过下调 PTEN 促进食管鳞状细胞癌进展和血管生成
作者还利用异种移植模型、肺转移模型和体内Matrigel栓血管生成实验验证岩藻糖基化外泌体来源的miR-6842-3p对癌症进展的影响(图6G-M)。如图6H-K所示,(miR-OE)岩藻糖基化外泌体显著促进ESCC生长、转移和血管生成,而过表达PTEN可挽救这些作用。一致地,IHC分析显示,异种移植模型中(miR-OE)岩藻糖基化外泌体组肿瘤组织中Ki-67和CD31表达显著升高(图6K)。然而,过表达PTEN可消除(miR-OE)岩藻糖基化外泌体介导的异种移植肿瘤组织中Ki-67和CD31的诱导表达(图6K)。在Matrigel栓血管生成实验中,与体外实验结果一致,miR-OE组Matrigel栓内血管结构更丰富。然而,过表达PTEN可消除miR-OE诱导的Matrigel栓内新生血管形成。综上,这些结果表明,ESCC细胞分泌的岩藻糖基化外泌体miR-6842-3p通过在体内靶向PTEN促进血管生成和ESCC进展。
7、岩藻糖基化外泌体miR-6842-3p通过激活AKT/mTOR信号通路促进食管鳞状细胞癌血管生成
PTEN作为一种公认的肿瘤抑制基因,调控多种细胞功能,包括细胞增殖和分化。在PTEN调控的信号级联反应中,AKT/mTOR通路是关键靶点,PTEN主要通过其内在的磷酸酶功能对其产生影响。为阐明肿瘤来源的岩藻糖基化外泌体miR-6842-3p促进ESCC进展和血管生成的分子机制,作者进行了转录组测序分析。转录组测序和京都基因与基因组百科全书(KEGG)富集分析显示,miR-6842-3p过表达下调的基因显著富集于8个关键通路,包括PI3K/AKT通路和趋化因子信号通路——基于这两个通路在促进肿瘤进展和血管生成中的关键作用,作者将其列为进一步研究的重点(图7A)。为进一步验证岩藻糖基化外泌体miR-6842-3p对PTEN/AKT/mTOR通路的调控作用,作者从稳定过表达或敲低miR-6842-3p的ESCC细胞系(KYSE150和TE-1)中分离岩藻糖基化外泌体,然后用这些外泌体处理HUVECs。Western blot结果显示,(miR-OE)岩藻糖基化外泌体下调HUVECs中PTEN蛋白水平,同时上调p-AKT和p-mTOR水平;相反,(miR-KD)岩藻糖基化外泌体处理显著降低p-AKT和p-mTOR水平,升高PTEN表达(图7B)。为明确(miR-OE)岩藻糖基化外泌体是否通过靶向PTEN激活AKT/mTOR信号通路,作者进行了挽救实验。结果显示,过表达PTEN可部分逆转(miR-OE)岩藻糖基化外泌体对HUVECs中p-AKT和p-mTOR表达的影响(图7C)。值得注意的是,AKT抑制剂伊帕替尼(Ipatasertib)可逆转(miR-OE)岩藻糖基化外泌体诱导的HUVECs中p-mTOR上调(图7D)。功能实验进一步证实,伊帕替尼或雷帕霉素(Rapamycin)处理可消除(miR-OE)岩藻糖基化外泌体对HUVECs的促增殖、促迁移和促血管生成作用(图7E-J)。综上,这些结果表明,岩藻糖基化外泌体miR-6842-3p通过下调PTEN表达并激活AKT/mTOR信号通路促进ESCC血管生成。
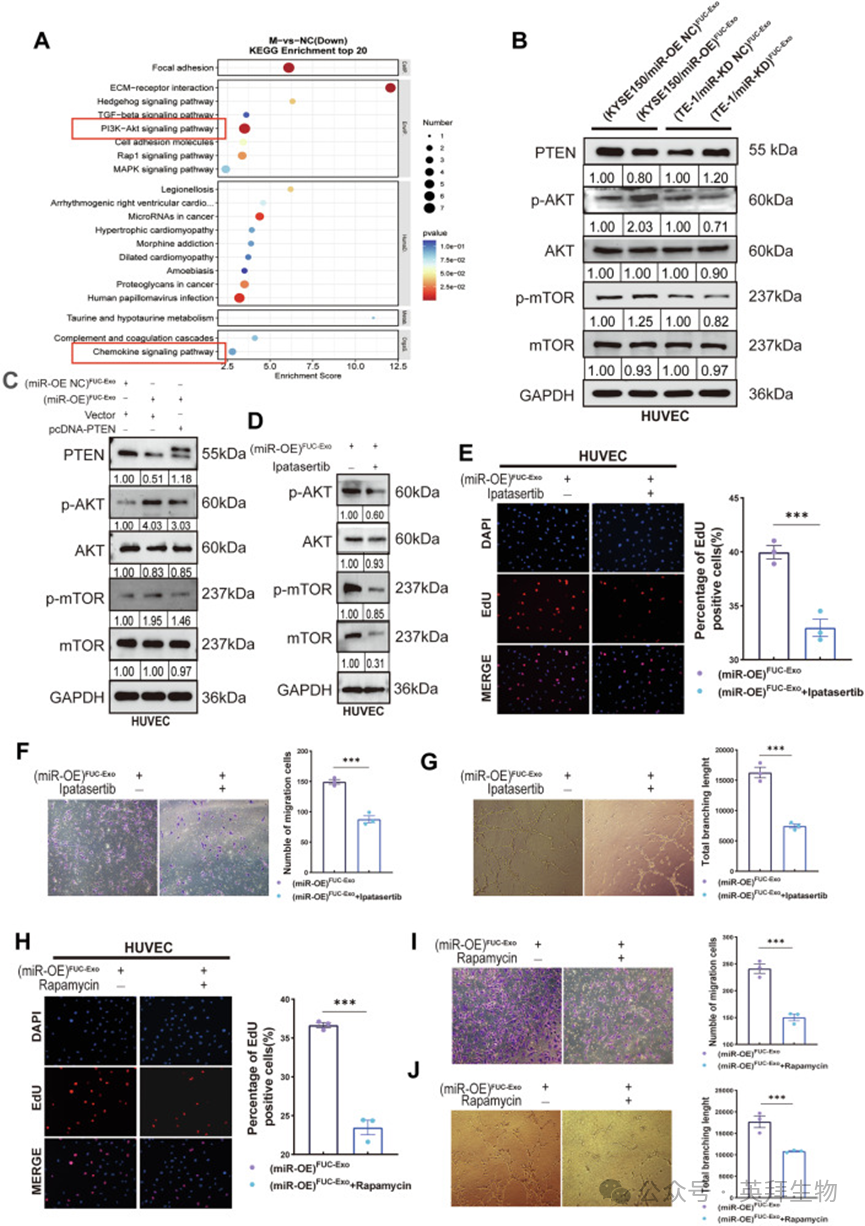
图7.岩藻糖基化外泌体 miR-6842-3p 通过激活 AKT/mTOR 信号通路促进食管鳞状细胞癌血管生成
8、岩藻糖基化外泌体miR-6842-3p通过AKT/mTOR信号通路下调CXCL10表达促进食管鳞状细胞癌血管生成
肿瘤微环境(TME)中的细胞因子在促进肿瘤进展和血管生成中发挥关键作用。作者的富集分析表明,趋化因子信号通路参与岩藻糖基化外泌体miR-6842-3p介导的致癌作用(图7A)。为进一步探究这一机制,作者用(miR-OE)岩藻糖基化外泌体或(miR-OE NC)岩藻糖基化外泌体处理HUVECs,通过人类细胞因子芯片分析细胞因子谱。岩藻糖基化外泌体处理后,CXCL10(也称为IP10)表达的显著降低引起了作者的关注(图8A)。CXCL10是一种已知的血管生成抑制剂,通过CXCR3依赖性和非依赖性途径发挥抗血管生成作用,包括抑制内皮细胞增殖、迁移和存活,以及诱导内皮细胞凋亡。CXCL10的抗血管生成特性已在多种癌症中得到广泛报道。例如,miR-21-5p通过抑制CXCL10表达促进ESCC血管生成。这些观察结果使作者推测,CXCL10可能是岩藻糖基化外泌体miR-6842-3p调控的AKT/mTOR信号通路的下游效应因子。
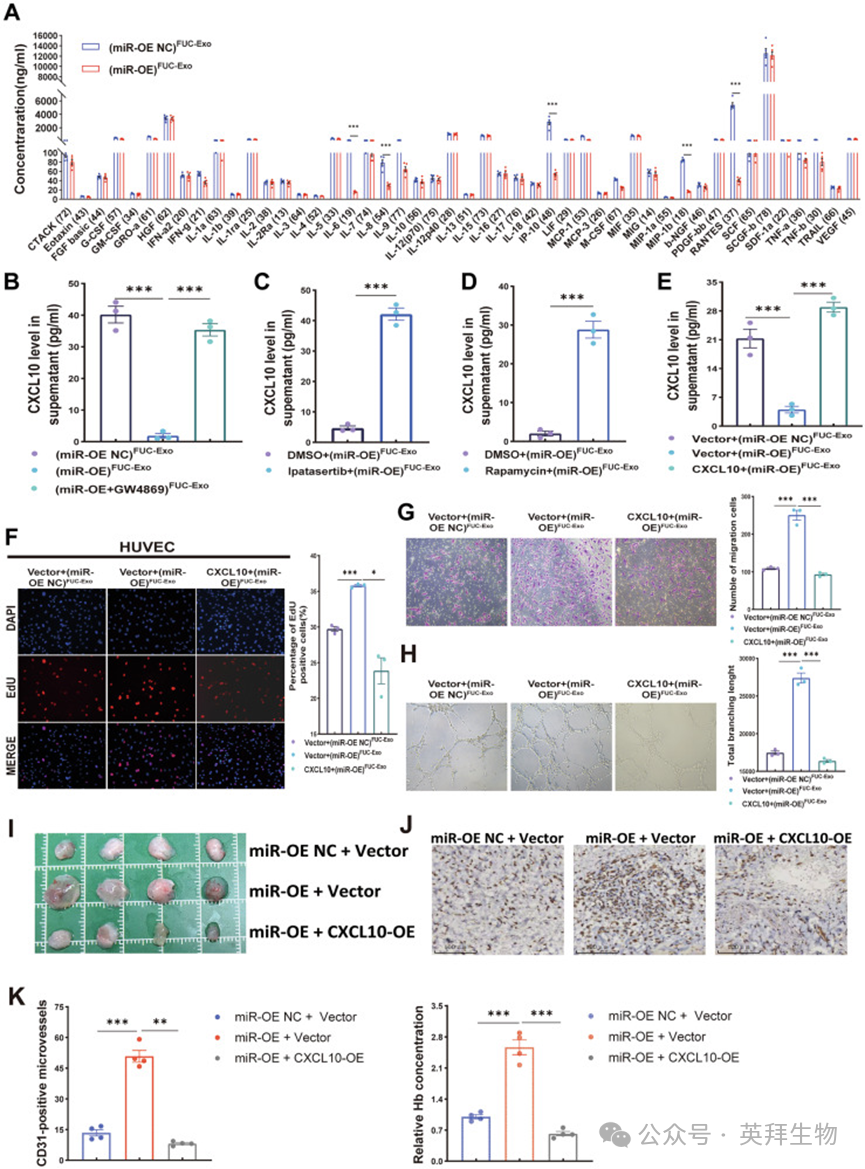
图8.岩藻糖基化外泌体 miR-6842-3p 通过调控 AKT/mTOR 信号通路下调 CXCL10 表达,进而促进食管鳞状细胞癌血管生成
随后的酶联免疫吸附测定(ELISA)实验显示,与对照组相比,(miR-OE)岩藻糖基化外泌体显著降低HUVECs中CXCL10的分泌(图8B)。相反,(miR-OE + GW4869)岩藻糖基化外泌体处理则增加CXCL10分泌(图8B)。此外,AKT抑制剂伊帕替尼和mTOR抑制剂雷帕霉素均能逆转(miR-OE)岩藻糖基化外泌体诱导的CXCL10下调(图8C、D)。同时,过表达CXCL10几乎完全挽救了(miR-OE)岩藻糖基化外泌体介导的HUVECs中CXCL10表达(图8E)。功能挽救实验进一步显示,过表达CXCL10显著减弱(miR-OE)岩藻糖基化外泌体的促血管生成作用,包括HUVECs增殖、迁移和管腔形成(图8F-H)。重要的是,体内Matrigel栓实验结果显示,过表达CXCL10也可逆转miR-6842-3p过表达在Matrigel栓中诱导的血管生成表型(图8I-K)。综上,这些结果表明,岩藻糖基化外泌体miR-6842-3p通过AKT/mTOR信号通路下调CXCL10表达,从而促进ESCC血管生成。
9、岩藻糖基化外泌体miR-6842-3p通过调控AKT/mTOR信号通路下调IRF1表达,进而抑制CXCL10产生
为阐明岩藻糖基化外泌体miR-6842-3p升高抑制CXCL10表达的机制,作者首先参考了相关前期研究。在肝细胞癌中,近期研究表明IRF1 mRNA水平与CXCL10呈正相关,并在CXCL10启动子区域鉴定出IRF1应答元件。此外,染色质免疫沉淀定量PCR(ChIP-qPCR)分析证实,IRF1结合可增强CXCL10转录,进而激活抗肿瘤免疫。值得注意的是,已有研究显示PI3K/AKT/mTOR信号通路的抑制也可通过IRF1上调CXCL10和CXCL11表达。基于这些发现,作者推测岩藻糖基化外泌体miR-6842-3p可能通过调控AKT/mTOR信号通路降低IRF1(CXCL10的转录调控因子)的表达,从而调控CXCL10的产生。
作者首先通过Western blot检测HUVECs中IRF1蛋白水平。结果显示,与对照组相比,(miR-OE)岩藻糖基化外泌体处理的细胞中IRF1表达显著降低,(miR-KD)岩藻糖基化外泌体处理的细胞中IRF1表达显著升高,证实(miR-OE)岩藻糖基化外泌体可下调HUVECs中IRF1表达(图9A)。鉴于PI3K/AKT/mTOR通路的抑制可通过IRF1上调CXCL10和CXCL11,作者进一步探究PTEN/AKT/mTOR通路是否在岩藻糖基化外泌体miR-6842-3p下游调控IRF1表达。挽救实验结果显示,过表达PTEN可部分逆转(miR-OE)岩藻糖基化外泌体对HUVECs中IRF1表达的影响(图9B)。此外,AKT抑制剂伊帕替尼或mTOR抑制剂雷帕霉素处理可消除(miR-OE)岩藻糖基化外泌体诱导的IRF1下调(图9C、D)。综上,这些结果表明,岩藻糖基化外泌体miR-6842-3p通过调控AKT/mTOR信号通路降低IRF1表达。
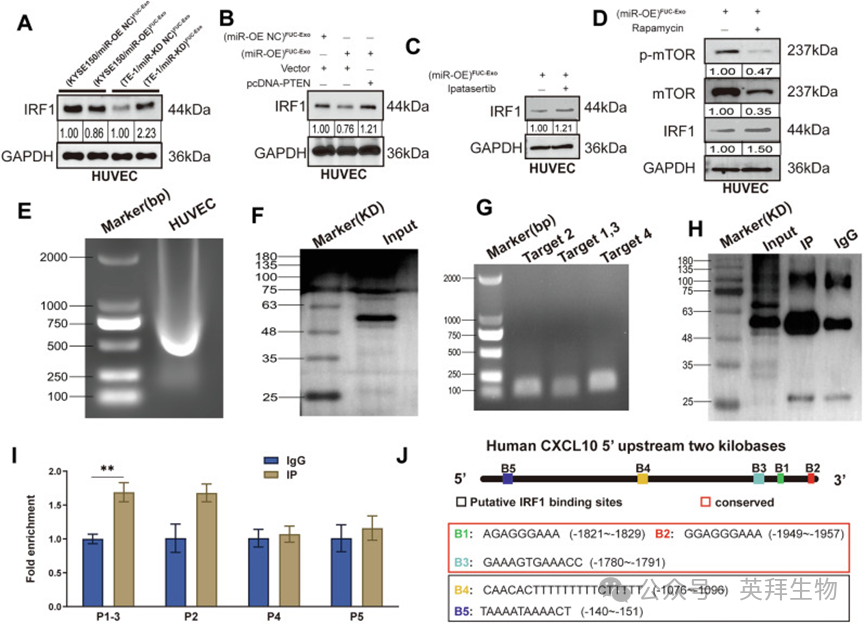
图9.岩藻糖基化外泌体miR-6842-3p通过调控AKT/mTOR信号通路下调IRF1表达,从而抑制CXCL10产生
鉴于IRF1作为调控CXCL10表达的主要转录因子,作者进一步探究IRF1是否在转录水平增强CXCL10表达。与前期研究一致,作者对癌症基因组图谱(TCGA)数据的分析显示,ESCC组织中IRF1与CXCL10表达呈显著正相关。为阐明IRF1介导的CXCL10调控的分子机制,作者首先利用生物信息学工具预测人类CXCL10启动子区域中的五个潜在IRF1结合基序。随后通过ChIP-qPCR验证IRF1在这些预测位点的结合情况。与免疫球蛋白G(IgG)对照组相比,ChIP-qPCR结果显示,在使用抗IRF1抗体进行免疫沉淀的HUVECs细胞裂解物中,IRF1在靶点1-3处的富集最强。值得注意的是,当使用抗IRF1抗体进行免疫沉淀时,在含有保守元件的区域检测到最高的IRF1富集(图9E-J)。综上,这些结果表明IRF1促进HUVECs中CXCL10转录。结合作者之前的结果,这证实岩藻糖基化外泌体miR-6842-3p通过下调IRF1表达抑制CXCL10产生,而这一过程是通过调控AKT/mTOR信号通路介导的。
参考文献
Chen J, Chen J, Lv X, Chen X, Ming L, Huang X, Wen F, Tang H, Gao Q, Liu C, Weng J, Huang Z, Zheng Y, Lin F, Chen W, Shang X, Yu C, Huang Y. Cancer-derived fucosylated exosomal miR-6842-3p as a novel marker promotes ESCC angiogenesis and metastasis via the PTEN/AKT/mTOR/IRF1/CXCL10 axis. Oncogene. 2026 Jan;45(1):104-120. doi: 10.1038/s41388-025-03643-2. Epub 2025 Nov 27. PMID: 41309934; PMCID: PMC12714579.