






PTC恶性进展新机制——代谢物乳酸通过组蛋白乳酸化改写表观遗传程序
甲状腺乳头状癌(PTC)是全球最常见的内分泌恶性肿瘤之一,其发病率稳步上升。虽然临床结局一般良好,但有临床意义的患者表现出高度侵袭性的肿瘤表型,其特征是肿瘤较大和淋巴结转移增加。越来越多的证据表明,代谢重编程和表观遗传失调是肿瘤进展的关键驱动因素。乳酸作为肿瘤代谢的副产物之一,近年来因其具有代谢以外的调节功能而受到关注。组蛋白乳酸化是一种受细胞内乳酸蓄积动态调控的表观遗传修饰,是肿瘤增殖、转移、免疫逃避和治疗抵抗的重要调节因子。然而,PTC中组蛋白乳酸化的功能意义和机制基础仍未被探索。在本研究中,我们发现PTC患者肿瘤组织中泛赖氨酸乳酸化和组蛋白H3赖氨酸18乳酸化(H3K18la)水平显著高于癌旁正常甲状腺组织。与侵袭性临床病理特征呈正相关。相关的细胞表型分析进一步支持这一结论。机制上,我们证明了H3K18la修饰直接促进了信号转导和转录激活因子1(STAT1)的转录激活。激活的STAT1继而促进乳酸脱氢酶A(LDHA)的转录上调,从而增强乳酸的生物合成并建立一个自我持续的正反馈回路。因此,肿瘤来源的乳酸通过“H3K18la-STAT1-LDHA”调节轴调控并维持PTC的恶性进展。综上所述,我们的发现揭示了PTC中肿瘤代谢和表观遗传调控之间的一种新的机制联系,为甲状腺癌的发病机制提供了重要的见解。此外,针对H3K18la-STAT1-LDHA轴的治疗可能是改善侵袭性和难治性PTC患者预后的一种创新和有前景的策略。该研究于2025年10月发表在《International Journal of Biological Sciences》,IF:10。
技术路线
主要研究结果:
1.组蛋白乳酸化在PTC中升高并与侵袭性肿瘤特征相关
我们的TMA分析显示,与肿瘤旁组织相比,PTC肿瘤组织中的泛赖氨酸乳酸化水平显著升高(p-tumor,图1A;图S1A中具有代表性的免疫组化染色)。
考虑到不同患者肿瘤(T)和淋巴结(N)分期的差异,我们根据染色强度将病例分为高和低乳酸化组。表现为高乳酸化水平的患者常表现为晚期T分期和侧颈部淋巴结转移(图1B和图1C)。
我们的研究结果表明,乳酰化可能在PTC中发挥促肿瘤作用,我们接下来试图在细胞水平上进一步研究这种可能性。我们评估了与正常甲状腺上皮细胞相比,PTC细胞系的糖酵解活性。PTC细胞表现出糖酵解率增加(图1D)、关键糖酵解酶(如己糖激酶2)的蛋白水平升高(图1E)、l -乳酸生成增加(图1F),从而增强组蛋白乳酸化(图1G)。
为进一步阐明可能参与PTC进展的特异性乳酸化标记,我们进行了靶向筛选。在候选修饰中,与正常对照相比,PTC细胞中的H3K18乳酸化(H3K18la)显著升高(图1H)。基于TMA的IHC分析证实,与p-肿瘤组织相比,肿瘤组织中的H3K18la表达显著较高(图1I), H3K18la升高与晚期T分期和颈部淋巴结转移相关(图1J和1K;图S1B)。此外,泛赖氨酸乳酸化水平与H3K18la水平呈正相关(图S1C)。此外,我们还检测了其他潜在的组蛋白乳酸化位点,如H3K9la和H4K12la,以及其他组蛋白修饰(H3K18ac和H3K18cr);其他乳酸化位点对PTC的恶性行为影响不大。事实上,其他组蛋白修饰甚至表现出一种矛盾的趋势,即较低的修饰水平与恶性进展增强相关。这一悖论可能反映了PTC生物学中复杂的表观遗传交互作用(图S1D-S1I,图S2A-S2F)。
综上所述,这些数据表明组蛋白乳酸化,尤其是H3K18la,可能促进PTC的恶性进展。
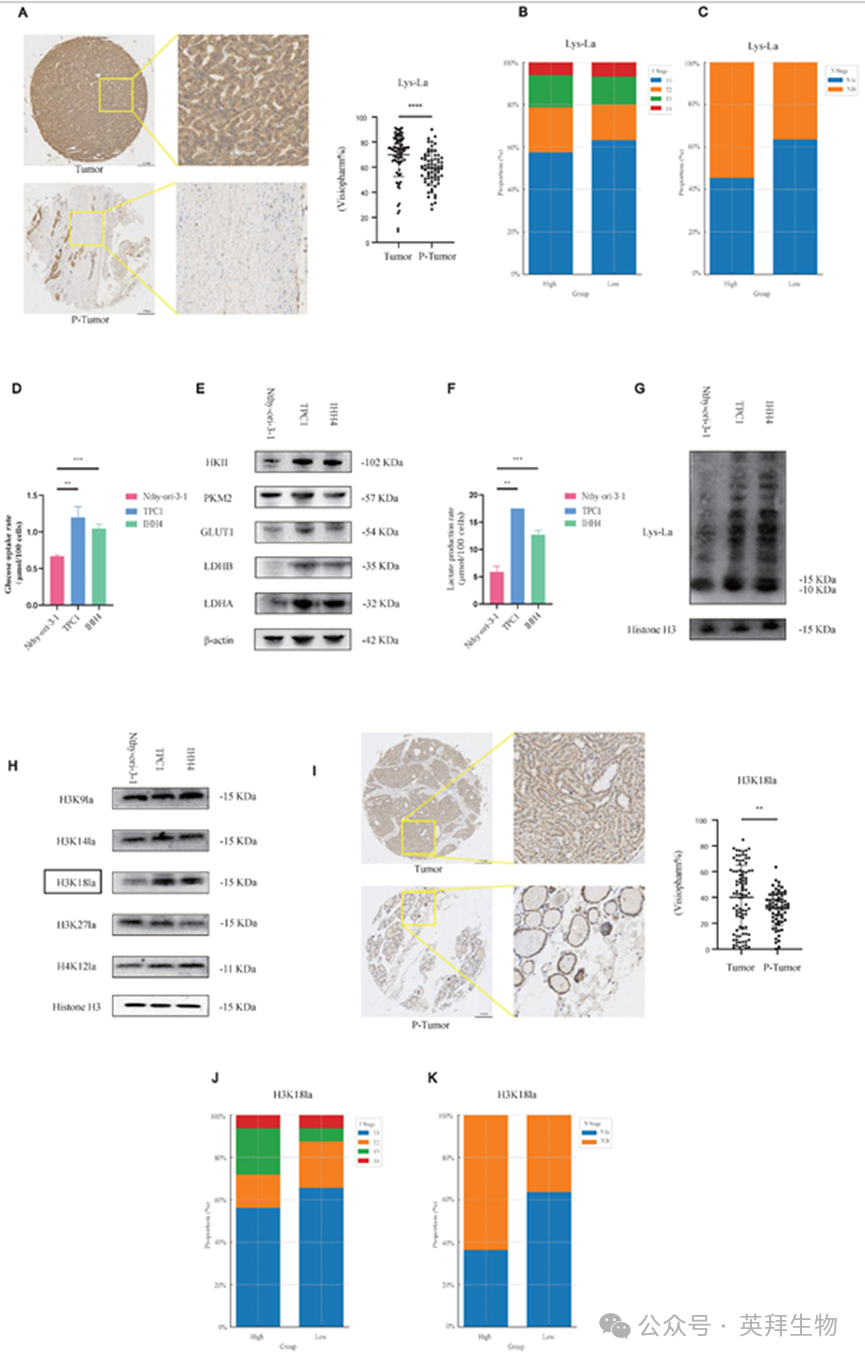
图1.组蛋白乳酸化在PTC中升高,H3K18la是最突出的位点,并与侵袭性肿瘤表型相关
2.组蛋白乳酸化调控PTC细胞增殖和侵袭
为研究组蛋白乳酸化在甲状腺乳头状癌中的功能,我们着重研究了H3K18la,它是之前发现的一个关键的乳酸化位点。我们使用2-DG(阻断葡萄糖-6-磷酸形成)和紫草素(A12125, AdooQ)(抑制丙酮酸激酶M2 (PKM2))来减少糖酵解通量和随后的组蛋白乳酸化(图2A)。
用增加浓度的2-DG或紫草素处理PTC细胞系后,组蛋白乳酸化和H3K18la特异性地出现剂量依赖性降低(图2B)。相应地,细胞增殖(图2C;图S3A)和侵袭能力(图2D和图2E;图S3B和S3C)显著受到抑制。此外,我们进一步研究了EP300是否介导了PTC中组蛋白的乳酸化。蛋白质印迹分析证实,敲低EP300导致H3K18la水平显著降低(图S3D)。此外,我们排除了糖酵解抑制剂(如2-DG)对EP300表达的任何直接影响(图S3E)。此外,鉴于Zhu等人的报告将KAT2A确定为潜在的组蛋白乳酸转移酶,我们在PTC中得出了相同的结论(图S3F)。
使用Nala进行的挽救实验部分恢复了2-DG处理诱导的受抑制的H3K18la水平和降低的乳酸生成速率(图2F和图S4A)。因此,Nala还部分恢复了PTC细胞的增殖(图2G;图4B)和侵袭(图2H和2I;图S4C和S4D),突显了乳酸介导的组蛋白乳酸化对肿瘤恶性程度的贡献。
体内实验中,我们使用IHH4细胞建立裸鼠皮下移植瘤。肿瘤达到约100 mm³后,小鼠被随机分为4组(图2J)。2-DG治疗显著抑制了肿瘤生长,而Nala可部分逆转这种抑制作用(图2K和2L;图S4E)。异种移植瘤的详细IHC染色结果见图S5A和S5B。
这些发现证实了组蛋白乳酸化(尤其是h3k18la)是PTC增殖和侵袭的关键调节因子,强调了其治疗潜力。
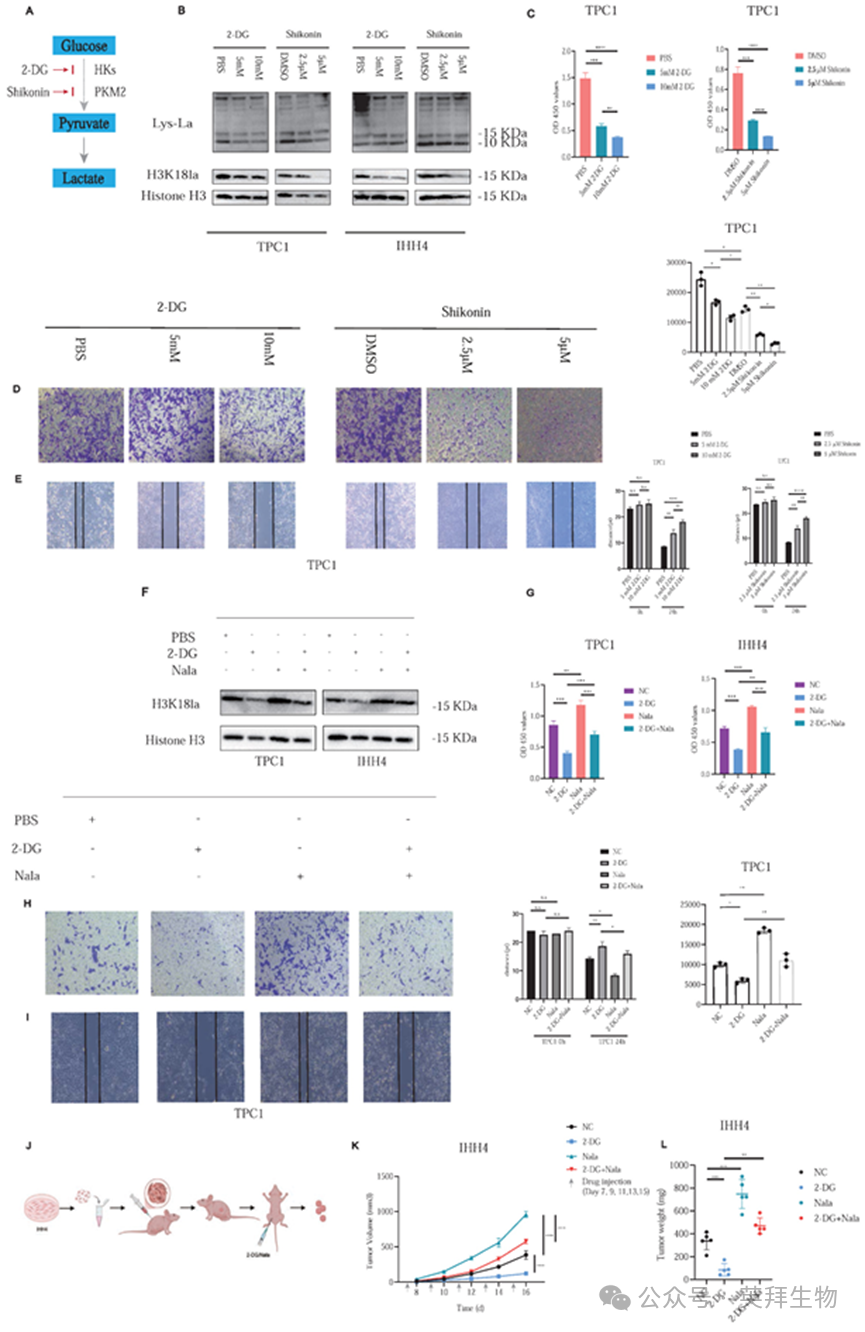
图2.组蛋白乳酸化调节PTC细胞的增殖和侵袭
3.乳酸积累导致H3K18la启动子区STAT1 RNA表达增强
为阐明组蛋白乳酸化对下游转录的影响,我们使用H3K18la特异性抗体进行了CUT&Tag分析,并结合后续的CUT&Tag-seq和RNA-seq分析。转录起始位点(TSSs)周围的H3K18la结合峰的分布密度表明,启动子区域显著富集(图3A-3C)。京都基因和基因组百科全书(KEGG)和基因本体论(Gene Ontology)富集分析表明,下调基因主要参与代谢通路,包括氧化磷酸化、碳代谢和HIF-1信号传导(图3D和3E)。
对3例匹配的PTC患者样本(GSE184362)的单细胞RNA-seq数据进行整合,确定了甲状腺上皮细胞中的差异表达基因(图3F)。结合cut - & tag -seq、RNA-seq、scRNA-seq和公开数据集,我们强调了STAT1是H3K18la的关键下游靶点(图3G;表S1)。TCGA和GTEx数据库分析证实,STAT1在甲状腺癌中上调(图3H)。
值得注意的是,CUT&Tag-seq显示STAT1启动子处显著的H3K18la富集(图4A)。基序分析确定了一个保守的结合位点(图4B),并由CUT&RUN-qPCR证实,这表明2-DG抑制糖酵解后,H3K18la结合减少(图4C)。荧光素酶检测显示,STAT1启动子活性可被Nala诱导,并被2-DG处理减弱,而这种作用在启动子突变时被消除(图4D;图S5C)。
2-DG和紫草素均降低了STAT1 mRNA(图4E)和蛋白水平(图4F), Nala部分挽救了这些降低(图4G)。此外,基于tma的IHC证实,肿瘤组织中的STAT1表达较高(图4H),与肿瘤大小(图4I)和颈部淋巴结转移(图4J;图S5D)增加相关。
这些发现强调了H3K18la介导的STAT1转录激活是促进PTC恶性的核心机制。
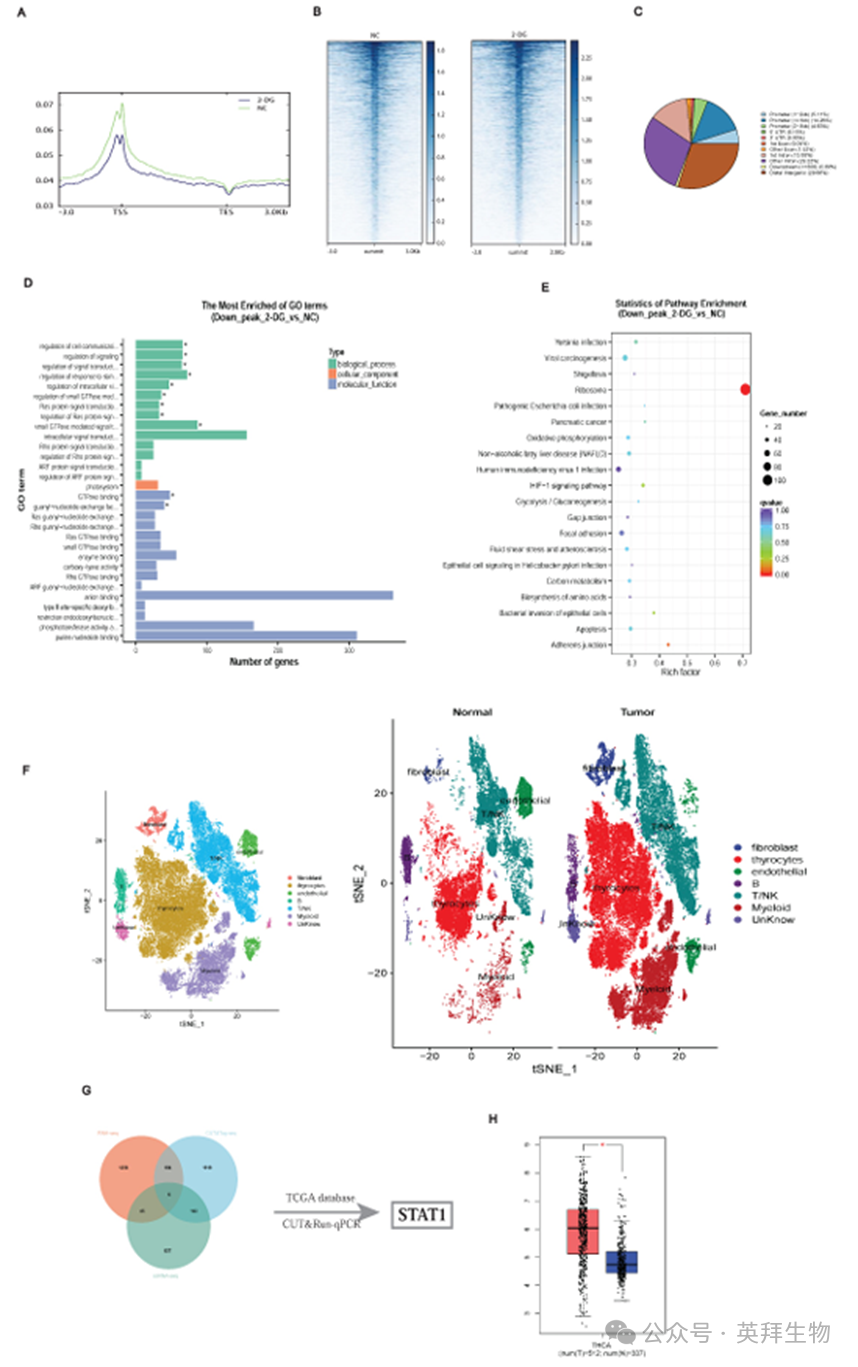
图3. H3K18la在PTC中潜在下游靶点的鉴定
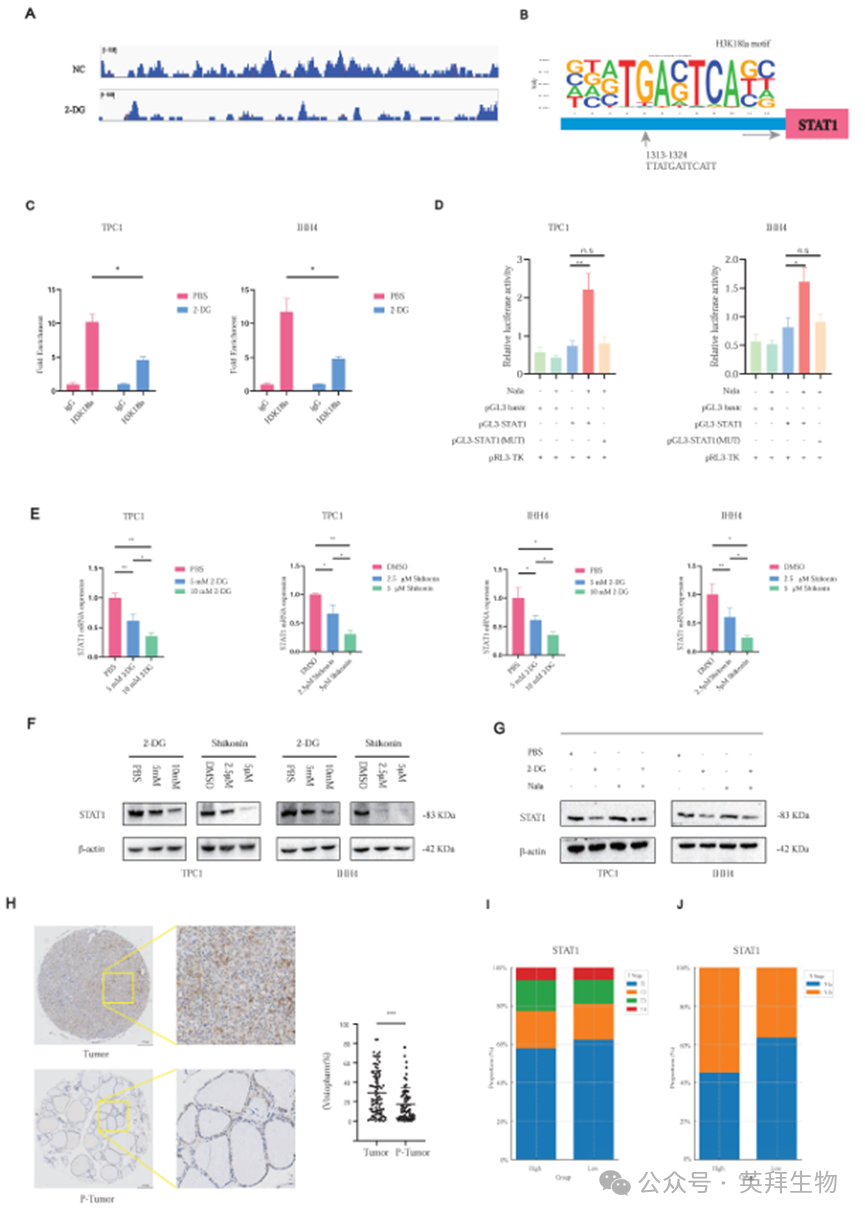
图4. H3K18la促进STAT1转录并与PTC的恶性特征相关
4.STAT1在PTC中作为细胞增殖和侵袭的致癌驱动因子
鉴于STAT1受到H3K18la的调控,我们建立了稳定的STAT1敲低(Sh-STAT1)和过表达(OE-STAT1) PTC细胞系(图5A和5F)。Nala处理部分恢复了Sh-STAT1细胞中受抑制的STAT1表达(图5B),相应地挽救了降低的增殖和侵袭(图5C-5E;图S6A-S6C)。相反,2-DG诱导的糖酵解抑制减弱了OE-STAT1细胞中增强的增殖和侵袭(图5G-5J;图S7A-S7C)。此外,基于TCGA和GTEx数据库的数据,以及PTC细胞系和正常甲状腺上皮细胞之间的比较分析,我们探索了其他STAT家族成员的表达模式,并发现在STAT家族中,只有STAT1在PTC中表现出促肿瘤作用(图S8A和S8B)。
在异种移植模型中,Sh-STAT1肿瘤显著较小,而OE-STAT1肿瘤相对于对照组表现出加速生长(图5K-5M;图S9A)。
总之,STAT1在PTC中起着致癌驱动作用,通过乳酸依赖的组蛋白乳酸化来调节恶性行为。
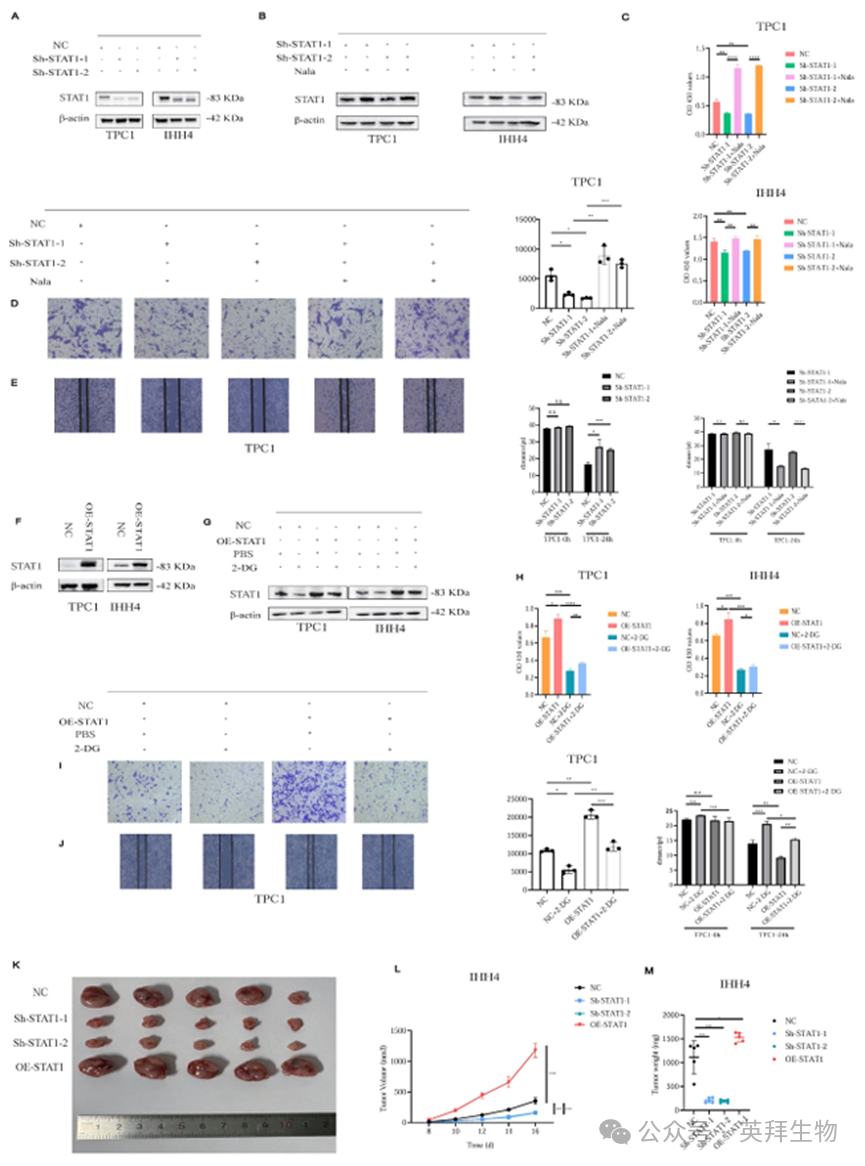
图5.H3K18la通过STAT1调控PTC细胞的增殖和侵袭
5.STAT1通过转录激活LDHA促进PTC乳酸生成
由于STAT1通常通过磷酸化(p-STAT1)起作用,我们验证了Sh-STAT1和e -STAT1细胞中p-STAT1的表达分别与STAT1水平相关(图6A和6B)。
为确定STAT1的转录靶点,我们将5个转录因子数据库与来自TPC1 Sh-STAT1和OE-STAT1细胞的RNA-seq数据整合(图6C;表S2)。KEGG分析显示代谢通路显著富集(图6D和6E)。值得注意的是,乳酸生成的限速酶LDHA被确定为STAT1的直接靶点。
CUT&RUN-qPCR验证了STAT1与LDHA启动子的结合,在Sh-STAT1细胞中显著降低,而在OE-STAT1细胞中增强(图6F和6G)。此外,鉴于在图1H中观察到的PTC细胞系中GLUT1和LDHB蛋白水平的增加,我们进行了额外的验证;然而,STAT1表达变化未能调节上述两种分子的转录(图S9B和S9C)。STAT1和LDHA的表达在mRNA(图6H)和蛋白水平(图6I)均呈正相关,TCGA分析进一步证实了这一点(图S9D)。AlphaFold3模型提示了STAT1和LDHA之间的潜在相互作用位点(图S9E)。
OE-STAT1细胞的l -乳酸生成显著增加,而Sh-STAT1细胞的l -乳酸生成则相反(图6J)。
我们的研究结果共同证明了一种正反馈机制,即肿瘤来源的乳酸促进H3K18la介导的STAT1转录,从而增强LDHA表达和乳酸生成。这一H3K18la-STAT1-LDHA轴在驱动PTC的增殖和转移中起关键作用,代表了一个新的治疗靶点(图7)。
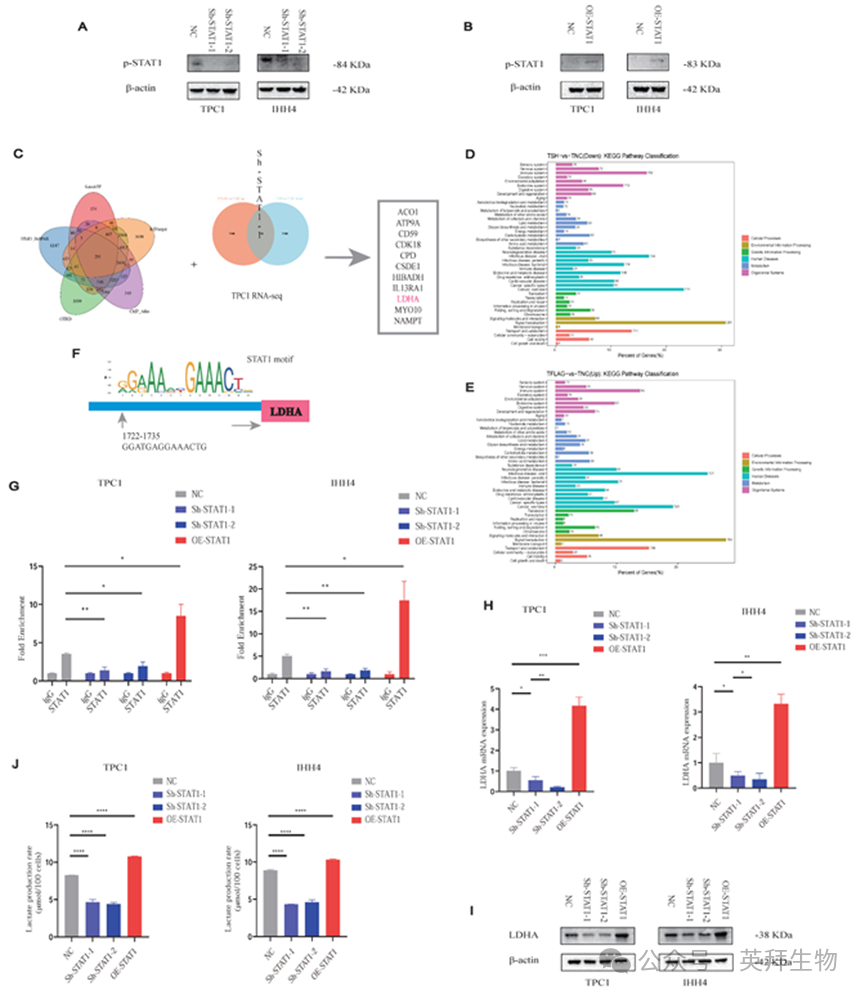
图6.乳酸通过H3K18la-STAT1-LDHA轴促进PTC恶性
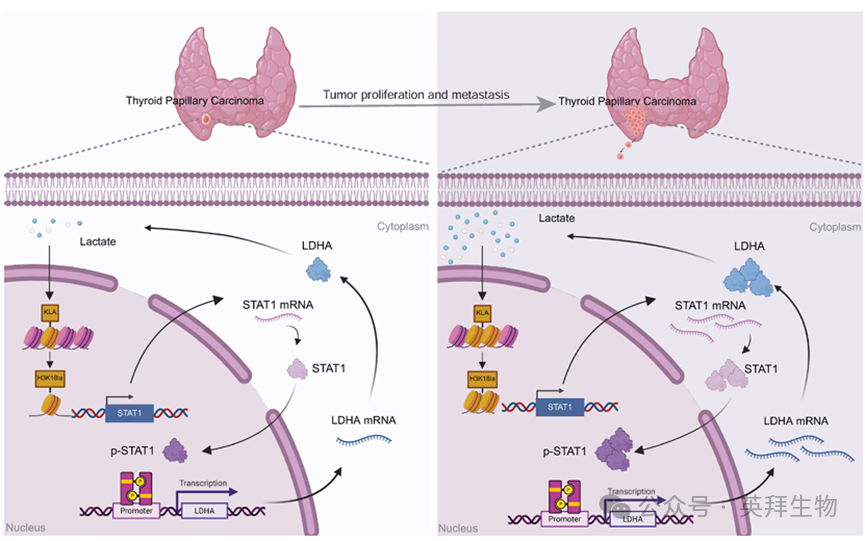
图7.提出的模型:肿瘤来源的乳酸增强H3K18la,H3K18la转录激活STAT1,从而促进LDHA的转录,增加其表达和乳酸生成。这形成了一个自我强化的H3K18la-STAT1-LDHA反馈回路,驱动PTC的恶性进展。
结论
综上所述,本研究全面描述了乳酸通过表观遗传调控驱动PTC恶性进展的分子机制。我们证明,肿瘤来源的乳酸通过激活H3K18la-STAT1-LDHA轴促进了一个自我维持的正反馈回路的建立,从而不断增加乳酸的产生,促进肿瘤的侵袭性。这些发现为了解癌症生物学中代谢-表观遗传相互作用的机制提供了新的见解,并为开发整合代谢重编程和表观遗传调控的治疗策略提供了坚实的理论基础。具体而言,针对H3K18la修饰和STAT1信号通路的药物靶点有望成为侵袭性和难治性PTC患者的治疗方法,具有巨大的临床转化潜力。
参考文献
Zhou Z, He C, Wang X, Jin X, Wen L, Yang Y, Zhou Q, Wang W, Teng L. Tumor-Derived Lactate Drives Malignant Progression of Refractory Papillary Thyroid Carcinoma via the H3K18la-STAT1-LDHA Axis. Int J Biol Sci. 2025 Oct 1;21(14):6373-6388. doi: 10.7150/ijbs.120277. PMID: 41208879; PMCID: PMC12594596.