






TRIM8 依赖的 K63 泛素化 PGK1 通过与 ACAT1 相互作用促进胃癌中的糖酵解和血管生成
糖酵解对促进癌症进展至关重要。然而,糖酵解调节血管生成过程的确切机制仍有待明确。本研究表明,在人胃癌细胞中,E3连接酶TRIM8促进糖酵解酶PGK1的k63连锁泛素化,并提高其稳定性,从而导致乙酰转移酶ACAT1募集,PGK1与ACAT1的相互作用增加,以及随后PGK1乙酰化依赖性糖酵解活性。该活性促进了PGK1介导的糖酵解、乳酸积累,并引发内皮细胞迁移和管形成显著增加,最终加速胃癌肿瘤血管生成。胃癌患者TRIM8水平与肿瘤血管生成及不良预后呈正相关。这些发现阐明了肿瘤细胞中K63泛素化调控糖酵解介导血管生成上调的新机制,并为靶向TRIM8依赖性PGK1 K63泛素化消除胃癌血管生成提供了分子基础。本文于2025年11月发表于Cell Death and Disease(IF=9.6)上。
技术路线:
结果:
1)TRIM8的表达与胃癌的进展和血管生成有关
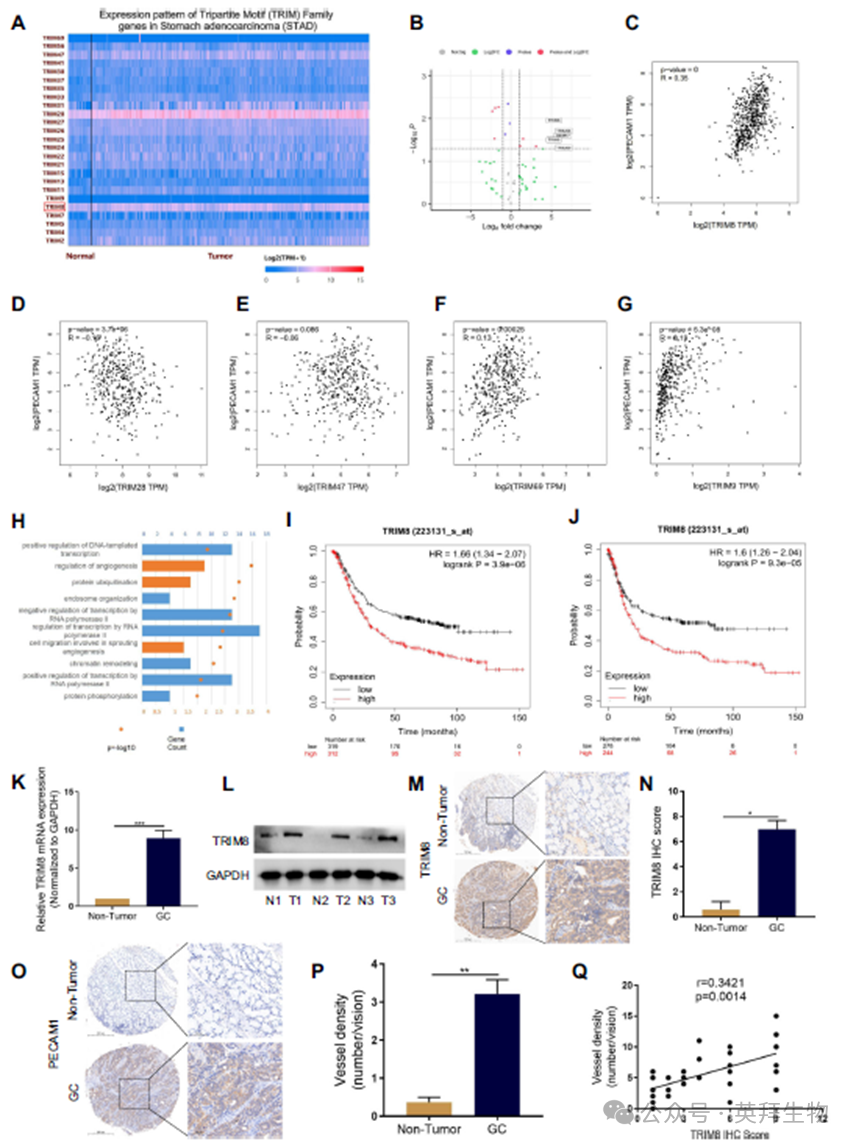
为了确定TRIM家族在胃癌中的潜在作用,我们首先使用TCGA数据库检测了TRIM家族在人胃癌组织和邻近非肿瘤组织中的表达水平。我们发现5个TRIM家族成员在GC肿瘤中的水平明显高于肿瘤周围组织(图1A,B)。值得注意的是,通过GEPIA数据库对TRIM家族成员和PECAM1在GC中的表达进行进一步的生物信息学分析,发现TRIM8的表达与GC组织的血管密度呈正相关(图1C-G)。对TCGA数据库中TRIM8相关基因的富集分析显示,与血管生成相关的生物过程中富集(图1H)。Kaplan-Meier Plotter分析显示,TRIM8表达与TCGA数据库中GC患者的生存和无进展生存呈负相关(图1I-J)。为了验证TRIM8与GC组织血管生成之间的相关性,我们测定了TRIM8和PECAM1在我们的GC患者队列中的表达模式。Western blot和免疫组化(IHC)染色显示TRIM8在肿瘤中的表达高于肿瘤周围组织(图1K-N)。同样,PECAM1在肿瘤中的表达也明显高于肿瘤周围组织(图1O-P)。综上所述,这些数据表明TRIM8表达上调与人胃癌血管生成呈正相关,预示着胃癌患者预后不良。
2)TRIM8促进GC血管生成
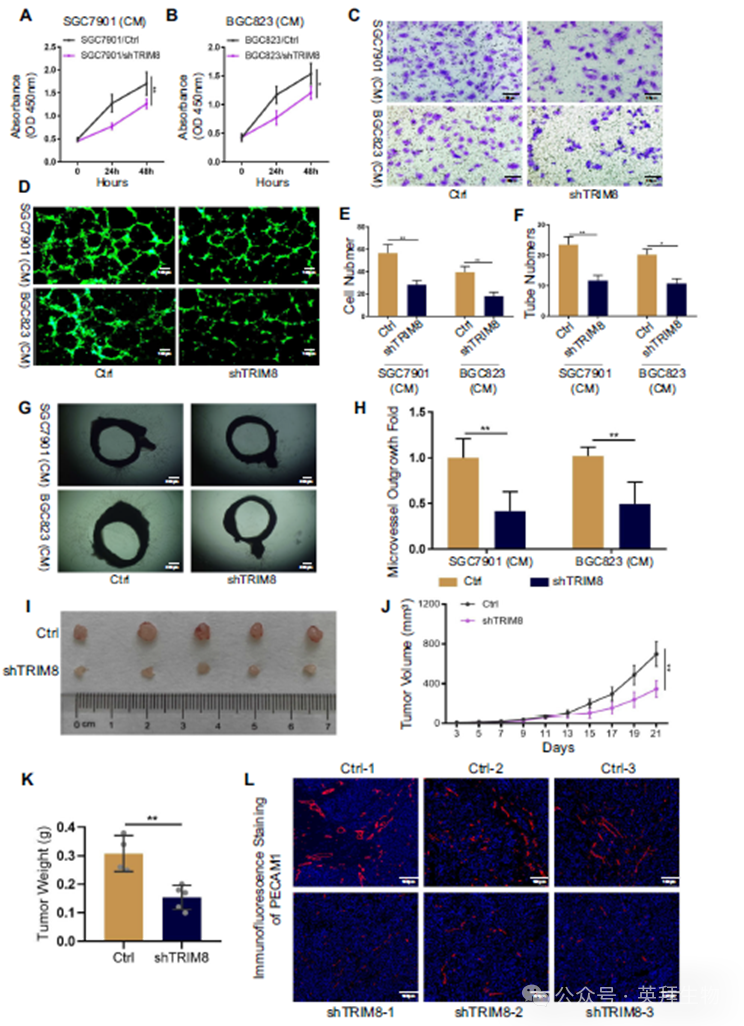
为了研究TRIM8在GC血管生成中的功能,我们利用质粒系统在两种常见的GC细胞系SGC7901和BGC823细胞中敲低TRIM8。来自SGC7901和BGC823细胞的条件培养基(CM)稳定地沉默TRIM8,降低了HUVEC的生长(图2A,B)。此外,Transwell实验证实,来自稳定的TRIM8沉默细胞系的CM减少了HUVEC的迁移(图2C,E)。肿瘤细胞可能通过改变内皮细胞的增殖和迁移来调节内皮细胞的形成。为了检验这种可能性,我们进行了内皮细胞管形成试验。与对照组相比,来自SGC7901/ shTRIM8和BGC823/shTRIM8细胞的CM引起HUVEC的管状形成明显减少(图2D,F)。接下来,我们在离体血管生成模型中研究TRIM8是否促进血管生长。采用野生型大鼠组织制备大鼠主动脉环。与对照GC细胞相比,添加SGC7901/ shTRIM8和BGC823/shTRIM8细胞系的CM后,在不添加其他生长因子的情况下,大鼠主动脉环的血管生长和分支明显减少(图2G,H)。我们通过异种移植小鼠模型进一步验证了TRIM8在GC中的体内促血管生成功能。与对照组小鼠相比,注射了TRIM8敲低GC细胞的小鼠肿瘤进展明显减慢(图2I-K)。PECAM1的IF显示,TRIM8的沉默导致肿瘤中血管密度降低(图2L)。综上所述,这些体外和体内的结果揭示了TRIM8在调节GC血管生成中的关键作用。
3)TRIM8促进K63连接的PGK1泛素化及其稳定性
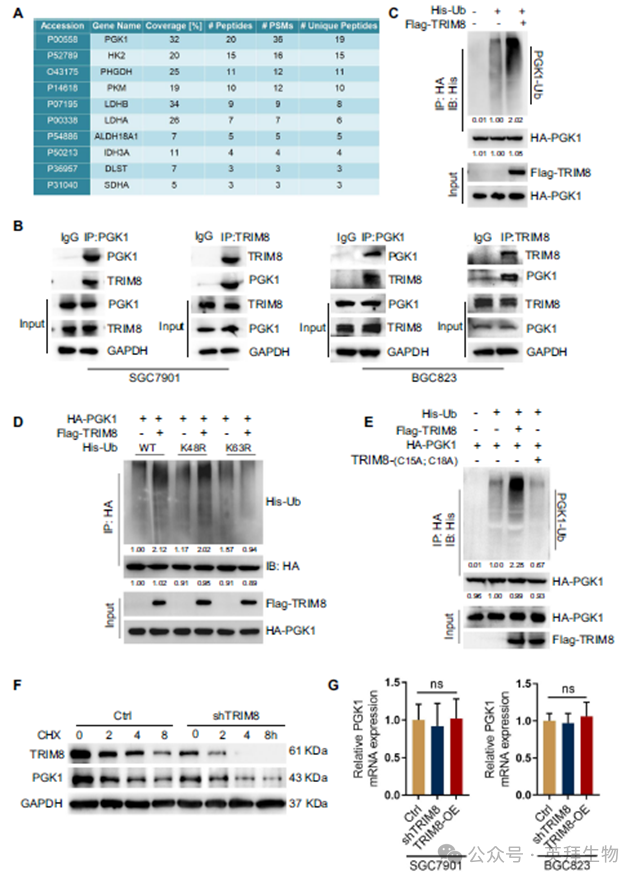
为了研究TRIM8介导血管生成的代谢相关机制,我们从SGC7901细胞中免疫沉淀TRIM8,并通过质谱分析分析TRIM8相关代谢蛋白。我们发现,糖酵解的主要代谢酶PGK1是一种与TRIM8相互作用的蛋白(图3A)。共免疫沉淀分析证实了这种相互作用,结果显示,在SGC7901和BGC823细胞中,内源性TRIM8与内源性PGK1相关(图3B)。此外,TRIM8 E3连接酶的异位表达容易诱导PGK1的泛素化(图3C)。这些结果表明TRIM8直接结合PGK1并促进其泛素化。K48和K63是最丰富的泛素连锁类型。野生型(WT)Ub和K63都可以通过TRIM8与PGK1连接,但K48不能。此外,TRIM8未能将K63R连接到PGK1(图3D)。这些数据表明TRIM8仅促进k63连接的PGK1泛素化。与这些发现一致的是,在翻译抑制剂环己亚胺存在的情况下,对PGK1衰变的分析显示,在治疗期间,TRIM8的缺失降低了PGK1蛋白的表达(图3F)。此外,TRIM8敲低和过表达均未能改变PGK1基因(mRNA)的表达(图3G)。TRIM8的RING结构域负责其E3连接酶活性。我们证明TRIM8,而不是TRIM8-(C15A; C18A) E3连接酶死亡突变体,在体外诱导PGK1泛素化(图3E)。综上所述,这些数据表明TRIM8是PGK1的直接E3连接酶,并促进其稳定性。
4)PGK1的K63泛素化促进GC细胞的糖酵解和血管生成
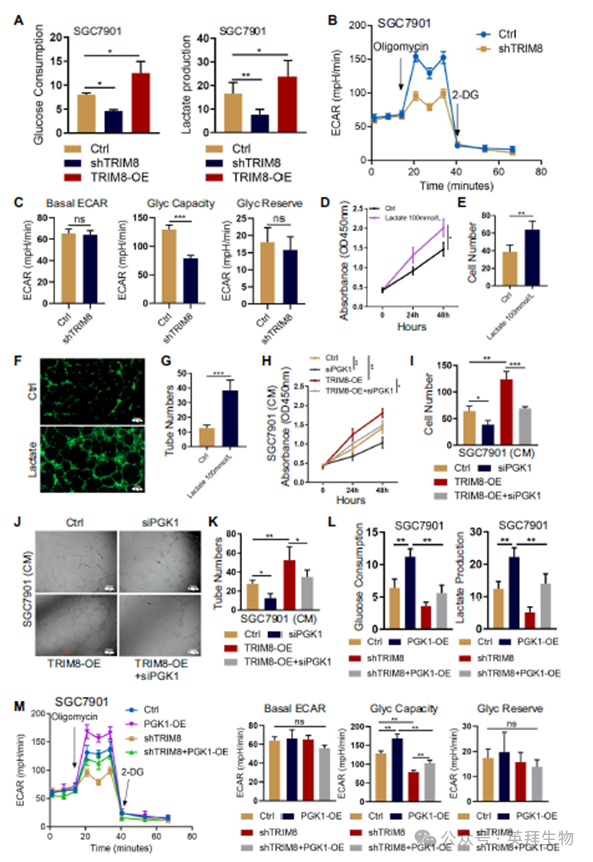
为了确定TRIM8介导的PGK1 k63泛素化的功能后果,我们研究了泛素化对GC细胞糖酵解的影响。我们发现PGK1-诱导的糖酵解在具有沉默TRIM8的GC细胞中比具有对照的GC细胞低得多,证明了较低的葡萄糖摄取和乳酸生成(图4A)。实时细胞外酸化速率(ECAR)进一步支持了这些结果,一致表明TRIM8缺失的GC细胞与对照细胞不同,无法促进糖酵解速率(图4B-C)。乳酸处理HUVECs促进内皮细胞生长和迁移,导致管形成增加(图4D-G)。为了确定PGK1 K63泛素化诱导的乳酸积累是否会影响肿瘤血管生成,我们进行了内皮细胞迁移和管形成实验。GC细胞中PGK1的敲低引发内皮细胞迁移和管形成的显著减少。PGK1沉默导致k63泛素化的PGK1减少,抵消了TRIM8过表达的促血管生成作用(图4H-K)。这些结果表明,K63泛素化对胃癌中PGK1促血管生成表型至关重要。值得注意的是,与对照相比,PGK1过表达具有更高的糖酵解活性,但它的过表达并没有抵消TRIM8沉默诱导的GC细胞糖酵解受损(图4L)。与这些观察结果一致,ECAR证实,沉默TRIM8可抑制PGK1过表达的能力,从而提高GC细胞的糖酵解率(图4M,N)。总的来说,我们的数据支持k63泛素化PGK1是TRIM8促血管生成作用的关键介质,可能还有其他因素参与TRIM8介导的糖酵解。
5)ACAT1介导的PGK1乙酰化促进PGK1活性,GC细胞糖酵解和肿瘤血管生成
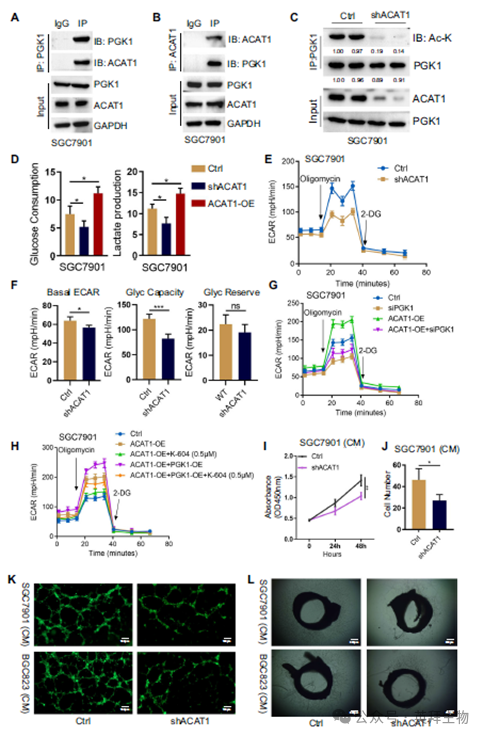
翻译后修饰是调节PGK1糖酵解酶活性的主要手段。质谱分析确定了一系列可能与TRIM8介导的PGK1糖酵解酶活性有关的乙酰基转移酶,包括ACAT1、NAA10、NAA15、DLAT和HAT1。免疫沉淀(IP)和Western blot显示ACAT1与PGK1相互作用(图5A,B),并促进PGK1乙酰化(图5C)。葡萄糖和乳酸检测显示,ACAT1基因敲低显著减少了GC细胞的糖酵解,葡萄糖消耗和乳酸生成的改变证明了这一点(图5D)。ECAR进一步支持了这些结果,一致表明ACAT1敲低能够降低GC细胞的糖酵解速率(图5E,F)。接下来,我们研究了ACAT1异位表达是否能够促进糖酵解,以及PGK1的乙酰化是否有助于这种作用。PGK1的沉默强烈抵消了ACAT1过表达促进糖酵解的作用(图5G)。为了证实ACAT1过表达促进糖酵解的作用是否依赖于PGK1的乙酰化,我们用有效的选择性ACAT1酶活性抑制剂K-604二盐酸预处理GC细胞。正如预期的那样,通过K-604处理,ACAT1过表达诱导的糖酵解促进作用被消除(图5H)。接下来,我们检测了GC细胞中ACAT1对肿瘤血管生成的影响。正如预期的那样,ACAT1缺失导致肿瘤血管生成减少,这可以通过内皮细胞增殖、迁移和管形成减少来检测(图5I-K)。与对照组相比,添加SGC7901/ shACAT1和BGC823/shACAT1细胞中的CM后,在没有添加其他生长因子的情况下,大鼠主动脉环的血管生长和分支明显减少(图5L)。这些结果表明,ACAT1介导的PGK1乙酰化是PGK1糖酵解酶活性、糖酵解和肿瘤血管生成所必需的。
6)ACAT1募集和PGK1乙酰化依赖于TRIM8介导的PGK1 K63泛素化
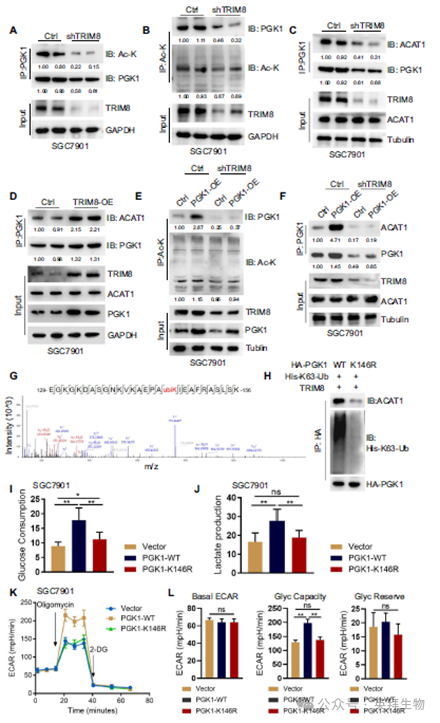
为了确定TRIM8介导的k63泛素化是否调节PGK1乙酰化,我们使用ac-赖氨酸抗体进行免疫印迹。Western blot检测PGK1的IP结果显示,在shTRIM8转染的SGC7901细胞中,ac-赖氨酸与PGK1蛋白的结合低于对照细胞(图6A)。Western blot后进行ac -赖氨酸富集显示,与对照细胞相比,shTRIM8-中的PGK1减少,与PGK1蛋白乙酰化程度降低一致(图6B)。机制上,在TRIM8敲低的GC细胞中,PGK1和ACAT1之间的内源性相互作用显著降低,而ACAT1蛋白水平不受TRIM8敲低的影响(图6C)。相比之下,在TRIM8过表达的GC细胞中,PGK1与ACAT1之间的内源性相互作用显著增加,而ACAT1蛋白水平不受TRIM8过表达的影响(图6D)。值得注意的是,GC细胞系中TRIM8的缺乏会在PGK1过表达时损害PGK1的乙酰化(图6E)。在TRIM8沉默的GC细胞中,PGK1与ACAT1过表达的内源性相互作用明显减少(图6F)。为了直接研究TRIM8介导的k63连接的PGK1泛素化在这些过程中的功能作用,我们接下来确定了PGK1上TRIM8依赖的泛素化位点。免疫沉淀的PGK1质谱分析显示,TRIM8在K146位点泛素化(图6G)。PGK1中K146R的突变也减少了TRIM8对PGK1的体外泛素化。在TRIM8存在的情况下,在共免疫沉淀实验中,缺乏K63连接的泛素化的PGK1也不与ACAT1相互作用(图6H),也不足以诱导GC细胞中PGK1糖酵解活性增强(图6I-L)。综上所述,这些数据表明TRIM8在K63连接PGK1 K146位点泛素化是ACAT1募集、PGK1乙酰化和PGK1糖酵解活性所必需的。
结论:
我们的研究结果强调了TRIM8通过K63泛素化调控的糖酵解在胃癌中的促血管生成作用,并提供了一种通过靶向TRIM8依赖的PGK1 K63泛素化来消除胃癌血管生成的潜在策略。
参考文献:
Feng A, Zhang J, Wang Z, Chen Z, Fang K, Li Z, Jiang H, Leng Z, Zhang S, Chu Y, Lian J, Chen T, Ye L, Xu M, He L. TRIM8-dependent K63-ubiquitinated PGK1 promotes glycolysis and angiogenesis in gastric cancer via interaction with ACAT1. Cell Death Dis. 2025 Nov 3;16(1):780. doi: 10.1038/s41419-025-08015-y.