






T细胞中TRIM25缺失介导的VISTA降解可增强癌症免疫治疗效果
当前免疫疗法的有限疗效凸显了对新靶点和联合治疗的迫切需求。V结构域T细胞活化抑制因子(VISTA)作为癌症免疫治疗中一个前景广阔的免疫检查点靶标,其调控机制尚不明确。通过CRISPR基因敲除筛选和蛋白质组学分析,作者发现含有三重基序的25蛋白(TRIM25)主要通过拮抗VISTA的降解信号,成为其正向调控因子。进一步研究表明,ERK介导的VISTA Thr284位点磷酸化过程可增强其与TRIM25的相互作用,从而稳定VISTA蛋白。VISTA衍生磷酸肽能竞争性破坏TRIM25-VISTA结合,进而降低VISTA表达水平,并显著增强PD-1/PD-L1阻断疗法的抗肿瘤效果。单细胞RNA测序分析显示,在T细胞特异性敲除Trim25的小鼠模型中,肿瘤浸润性CD8+ cytotoxic T细胞数量显著增加。值得注意的是,T细胞中Trim25的基因缺失不仅能改善抗PD-L1免疫治疗效果,还能在不同小鼠肿瘤模型中显著增强CAR-T细胞的抗肿瘤活性。总之,作者的研究系统揭示了T细胞中VISTA的调控机制,并强调靶向TRIM25-VISTA轴可作为增强肿瘤免疫治疗的潜在策略。该研究于2025年11月发表在《Cell Research》,IF 25.9分。
技术路线:
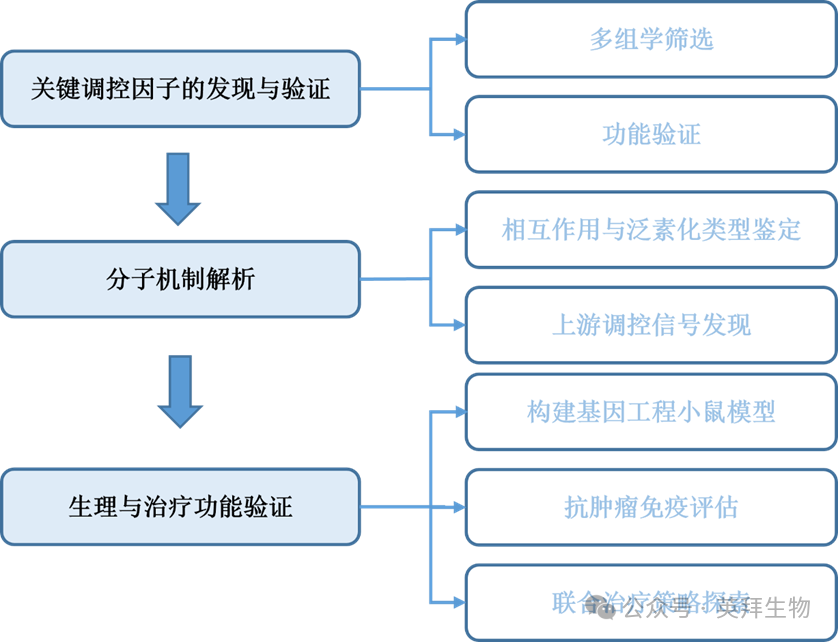
主要研究结果:
1)CRISPR基因敲除筛选与蛋白质组学分析鉴定TRIM25为VISTA蛋白稳态的关键调控因子
泛素化是一种重要的翻译后修饰,可调控蛋白质稳态、亚细胞定位及蛋白质-蛋白质相互作用。由泛素E3连接酶或去泛素化酶介导的异常泛素化会导致包括癌症在内的多种人类疾病。为鉴定VISTA蛋白表达的关键调控因子,作者采用靶向泛素化相关蛋白的CRISPR基因敲除文库(包含10,108条靶向660个基因的sgRNA及1000条非靶向对照sgRNA)进行了体外筛选。在稳定表达C端GFP标签VISTA(VISTA-GFP)的HEK293T细胞中高效转导sgRNA文库后,通过荧光激活细胞分选术(FACS)基于GFP信号强度进行分选(图1a上图)。为识别调控VISTA蛋白表达的基因,作者收集了GFP信号最低20%(GFP低表达)与最高20%(GFP高表达)的细胞群体(图1a上图)。测序结果显示所有样本的sgRNA文库覆盖率接近100%,且基尼指数较低,表明sgRNA丰度与分布符合要求且无偏倚(图1a)。进一步分析鉴定出109个VISTA蛋白表达的潜在调控基因(图1a上图)。作为补充策略,作者通过免疫共沉淀(IP)联合质谱(MS)分析鉴定了VISTA相互作用蛋白(图1a下图),共发现488个潜在VISTA结合蛋白(图1a下图)。通过对比CRISPR筛选与蛋白质组学分析结果,作者鉴定出三个在两项实验中共同出现的泛素E3连接酶候选因子:TRIM25、UBR5与KLHL12(图1a),提示这三种蛋白可能参与调控VISTA蛋白稳态。
为确定哪种泛素E3连接酶对VISTA蛋白丰度的调控效能最强,作者通过shRNA敲低TRIM25、UBR5或KLHL12并评估其对VISTA蛋白表达的影响。作者首先分析了多种癌细胞系(包括造血系统肿瘤与实体瘤)中VISTA的表达水平。与主要在实体瘤细胞系中表达的免疫检查点PD-L1和B7-H3不同,VISTA主要表达于造血系统肿瘤细胞系,部分实体瘤细胞系亦有表达。值得注意的是,在人白血病T细胞系Jurkat和MOLT-4中VISTA表达水平较高。据此,作者评估了敲低这些E3连接酶对Jurkat和MOLT-4细胞中VISTA蛋白水平的影响。结果显示,在这两种细胞系中敲低TRIM25(而非UBR5或KLHL12)可显著降低VISTA蛋白水平,且不引起VSIR mRNA水平的明显变化(图1b-e),提示TRIM25可能在翻译后水平稳定VISTA。鉴于VISTA为膜蛋白,作者通过流式细胞术检测敲低TRIM25是否降低Jurkat细胞表面VISTA水平,结果证实TRIM25敲低可显著减少细胞表面VISTA蛋白(图1f,g)。一致地,细胞组分分离实验在Jurkat和MOLT-4细胞中均证实,敲低TRIM25后膜组分中VISTA蛋白显著减少。此外,异位表达野生型TRIM25(而非酶活性失活的TRIM25 C50/53S突变体[将第50和53位半胱氨酸均突变为丝氨酸])可大幅提升VISTA蛋白丰度而不改变其mRNA水平(图1h,i),表明TRIM25的泛素E3连接酶活性是其正向调控VISTA蛋白水平所必需的。
进一步地,作者利用Actinomycin抑制蛋白质翻译并检测VISTA蛋白半衰期。野生型TRIM25显著延长VISTA蛋白半衰期,而酶失活突变体TRIM25 C50/53S无此效应(图1j,k)。相反,敲低TRIM25缩短了Jurkat细胞中VISTA的蛋白半衰期(图1l,m)。这些结果进一步证实TRIM25主要在翻译后水平稳定VISTA。
为探究临床相关性,作者采用多重免疫组织化学染色评估了人结直肠癌样本中TRIM25与VISTA表达的相关性。首先使用稳定表达shTRIM25、shVSIR或对照shGFP的Jurkat细胞验证了TRIM25与VISTA抗体的免疫组化特异性。经验证后的mIHC分析显示,在人结直肠癌组织肿瘤微环境的CD3+ T细胞中,TRIM25与VISTA存在显著共定位,提示病理状态下两者表达呈正相关(图1n)。为进一步探索TRIM25-VISTA轴与结直肠肿瘤患者临床预后或治疗结局的关联,作者对接受免疫治疗患者的标本进行IHC染色,发现相较于治疗应答者,抗PD-1治疗无应答者肿瘤组织中TRIM25与VISTA水平显著升高,提示TRIM25-VISTA轴可能在临床环境中介导免疫治疗耐药。综上所述,这些结果表明TRIM25可能通过正向调控T细胞中VISTA蛋白稳定性促进肿瘤免疫逃逸。
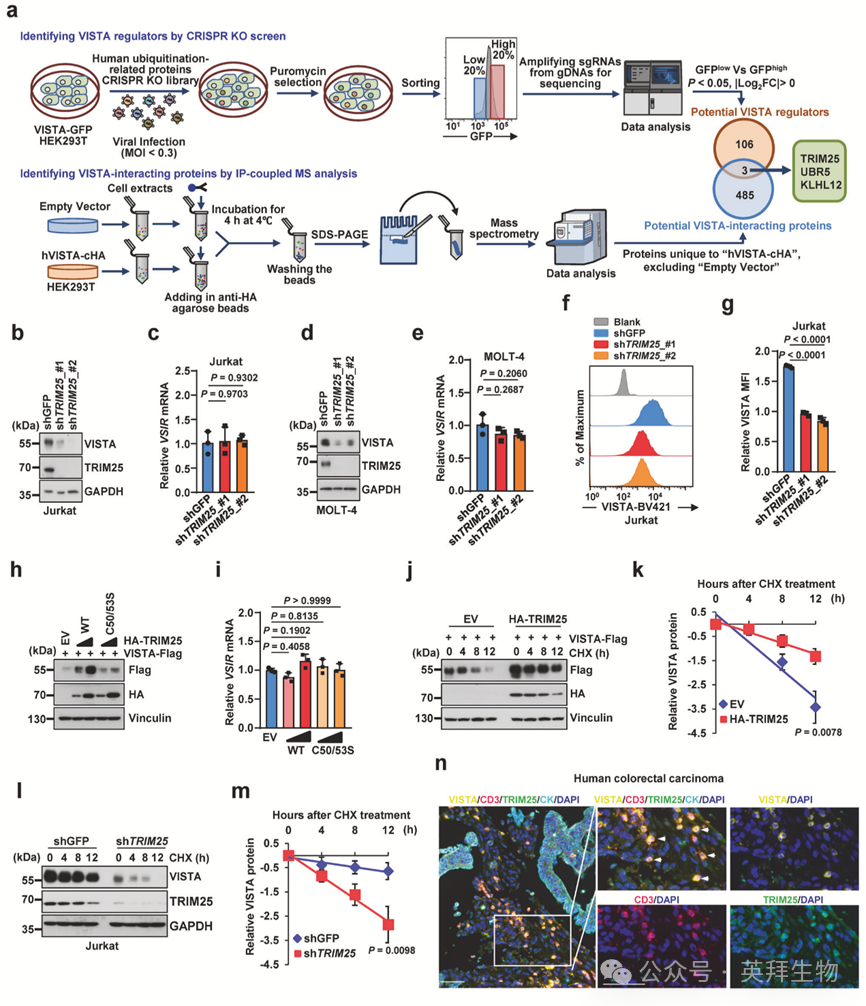
图1:CRISPR敲除筛选与蛋白质组学分析鉴定TRIM25为VISTA蛋白稳态的关键调控因子
2)TRIM25与VISTA相互作用并促进其K63连接型泛素化
作者的IP-MS结果表明TRIM25是细胞内VISTA的潜在结合伴侣(图1a下图)。作者首先通过免疫共沉淀实验证实TRIM25在Jurkat细胞中与VISTA存在相互作用(图2a)。GST pull-down实验也验证了TRIM25与VISTA蛋白间的相互作用(图2b)。由于TRIM25包含N端RING结构域、卷曲螺旋结构域和C端SPRY结构域,作者探究了TRIM25的哪个结构域是与VISTA相互作用所必需的。为此,作者构建了TRIM25的多种截断突变体并检测其与VISTA的结合能力(图2c)。结果显示,缺失C端SPRY结构域(Δ439–630,对应去除第439至630位氨基酸)会完全破坏与VISTA的结合,而缺失N端RING结构域或卷曲螺旋结构域则无此影响,表明TRIM25的SPRY结构域对于其与VISTA相互作用至关重要(图2c-e)。此外,缺失胞质结构域的VISTA突变体(Δ216–311,对应去除第216至311位氨基酸)丧失了与TRIM25的结合能力(图2f,g),提示VISTA通过其胞质结构域与TRIM25相互作用。
泛素能够在其靶标蛋白上形成多种连接方式的泛素链,从而决定蛋白质的最终命运。学界公认K48连接型泛素链可作为26S蛋白酶体介导蛋白质降解的信号。相比之下,K63连接型泛素化作为非降解信号,在某些情况下调控蛋白质-蛋白质相互作用或拮抗K48连接型泛素化以增强蛋白质稳定性。既往研究表明TRIM25既可作为泛素E3连接酶,也可作为类泛素干扰素刺激基因15(ISG15)连接酶,分别主要介导底物的K48连接型、K63连接型泛素化或ISG化修饰。因此作者探究了TRIM25主要催化VISTA的哪种修饰类型。结果显示VISTA被泛素化显著修饰,但未检测到ISG化修饰(图2h)。此外,异位表达TRIM25可促进VISTA的泛素化(图2h)。为探索TRIM25可在VISTA上组装哪种类型的泛素链,作者将仅保留特定赖氨酸的泛素突变体(K6O、K11O、K27O、K29O、K33O、K48O和K63O,这些突变体仅保留指定赖氨酸位点,同时将泛素分子上其余六个赖氨酸均突变为精氨酸)分别与VISTA共转染至HEK293T细胞,并进行体内泛素化实验。结果显示VISTA主要被K48和K63连接型泛素化修饰,同时也可检测到K11和K27连接型泛素化(图2i)。然而TRIM25显著促进VISTA的K63连接型(而非K48连接型)泛素化(图2j)。TRIM25-C50/53S失活突变体丧失了增强VISTA K63连接型泛素化的能力(图2k)。在Jurkat细胞内源性VISTA蛋白水平上也观察到了K63连接型泛素化修饰。
有趣的是,增加K63O泛素表达会逐步抑制VISTA的K48连接型泛素化。此外,异位表达TRIM25在增强K63连接型泛素化的同时降低了VISTA的K48连接型泛素化。当与泛素K63R突变体(仅将泛素分子第63位赖氨酸变为精氨酸)共转染时,TRIM25无法再促进VISTA K63连接型泛素化,进一步支持TRIM25主要在细胞内催化VISTA的K63连接型泛素化。与这些观察一致,敲低TRIM25可降低内源性VISTA的K63连接型泛素化并增加其K48连接型泛素化(图2l)。这些发现共同提示TRIM25通过促进VISTA的K63连接型泛素化来拮抗K48连接型泛素化的降解效应,从而稳定VISTA。
由于TRIM25主要是与VISTA胞质尾端(第216-311位氨基酸)相互作用的胞质E3连接酶,作者推测TRIM25介导的K63连接型泛素化发生在此区域内。序列分析显示在人和小鼠VISTA的胞质尾端存在两个保守赖氨酸残基——K217和K258。为确定K63连接型泛素化的主要位点,作者在这些位点构建了赖氨酸至精氨酸点突变体。体内泛素化实验显示,异位表达TRIM25显著增强野生型VISTA和K258R突变体的K63连接型泛素化,但对VISTA的K217R突变体无此作用。此外,在TRIM25存在的情况下,与野生型VISTA相比,K217R突变体表现出K63连接型泛素化减少和K48连接型泛素化增加。一致地,TRIM25过表达可提高野生型VISTA的蛋白水平,但对K217R突变体的稳定性无影响。无论是野生型TRIM25还是其催化突变体均不能增强VISTA K217R突变体的蛋白丰度。进一步支持这一发现,Actinomycin追踪实验显示在表达TRIM25野生型与C50/53S突变体的细胞中,K217R突变体的半衰期无显著差异。这些结果共同鉴定出K217是TRIM25介导VISTA K63连接型泛素化的主要位点,该修饰对稳定VISTA至关重要。
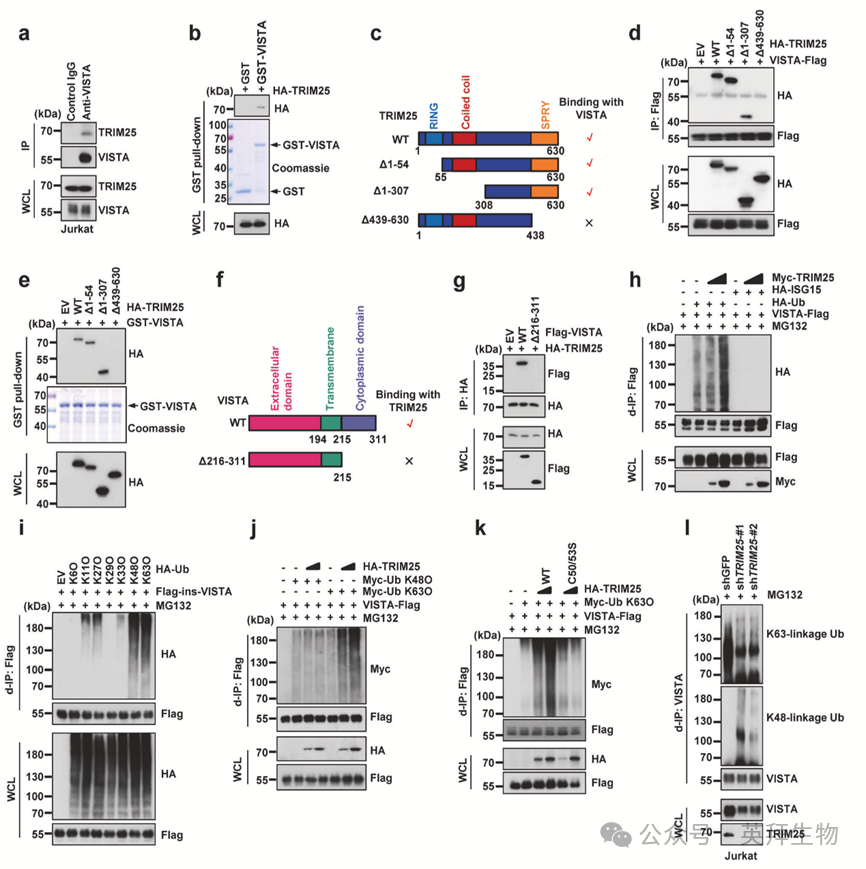
图2:TRIM25与VISTA相互作用并促进其K63连接型泛素化
3)14-3-3ε识别ERK介导的VISTA Thr284磷酸化,促进VISTA与TRIM25的相互作用
14-3-3家族蛋白作为分子衔接蛋白,能够识别蛋白质上的磷酸丝氨酸/磷酸苏氨酸残基并调控蛋白质-蛋白质相互作用。通过分析IP联合MS的结果,作者发现14-3-3ζ和14-3-3ε均被鉴定为VISTA的潜在相互作用伴侣(图3a)。为确定14-3-3家族蛋白中对VISTA具有强亲和力的特定亚型,作者进行了免疫共沉淀实验。值得注意的是,14-3-3ε与VISTA的相互作用尤为显著,优于其他14-3-3亚型(如β、γ、σ和ζ)(图3b)。为探究磷酸化事件对VISTA与14-3-3ε相互作用的影响,作者在共沉淀反应体系中加入λ磷酸酶处理。λ磷酸酶处理显著削弱了VISTA与14-3-3ε之间的相互作用(图3c),提示VISTA的磷酸化状态有助于其与14-3-3ε的结合。此外,VISTA Δ216–311突变体无法与14-3-3ε相互作用(图3d),表明VISTA的C端胞质结构域负责与14-3-3ε结合。
有趣的是,作者发现异位表达14-3-3ε可增强VISTA与TRIM25的结合。为确定14-3-3ε、VISTA和TRIM25是否形成三元复合物,作者进行了串联免疫共沉淀实验,结果证实这些蛋白间可形成稳定复合物(图3e)。在Jurkat细胞中进行的共沉淀实验也在内源性水平验证了14-3-3ε、VISTA和TRIM25的相互作用(图3f)。重要的是,敲低14-3-3ε显著降低了VISTA与TRIM25的结合,进一步支持14-3-3ε在促进该复合物形成中的关键作用(图3g)。这些发现共同证明14-3-3ε在促进VISTA与TRIM25结合中发挥着关键支架功能。
由于14-3-3ε通常作为磷酸丝氨酸/苏氨酸模体的阅读器,且λ磷酸酶处理显著削弱了VISTA与14-3-3ε的相互作用(图3c),作者推测VISTA磷酸化促进其与14-3-3ε的结合。为鉴定负责VISTA磷酸化的激酶,作者使用SCANSITE 4.0数据库进行了生物信息学分析,预测出两个推定的ERK1磷酸化位点(PST284P)。此外,将VISTA D结构域中四个保守基序残基突变为丙氨酸,会破坏其在细胞内与ERK1的相互作用。
体外激酶实验进一步证明,在相同条件下ERK1能有效磷酸化GST标签的VISTA,但不能磷酸化单独的GST标签(图3i)。而且,在VISTA C端截短突变体ΔC277–311和ΔC245–311中,ERK1介导的磷酸化信号消失(图3i),提示关键的ERK1磷酸化位点可能位于第277-311位氨基酸区间。在潜在位点中,Thr284突变为丙氨酸会基本消除磷酸化,而Ser287和Ser305的突变则无此效应,由此确定Thr284为主要磷酸化位点(图3j)。
为研究位点特异性磷酸化,作者制备了靶向VISTA Thr284磷酸化的特异性抗体。斑点印迹分析证实该抗体能特异性检测Thr284磷酸化肽段,且不与未磷酸化对应肽段发生交叉反应。与这些结果一致,pT284-VISTA抗体选择性识别野生型VISTA,但不能检测T284A磷酸化缺陷突变体,该突变体也丧失了与14-3-3ε结合的能力(图3k,l)。为探究ERK与VISTA磷酸化的关系,作者检测了调控ERK活性后的pT284-VISTA水平。异位表达ERK激酶显著增加了表达野生型VISTA细胞中的pT284-VISTA信号,但对T284A突变体无此作用(图3m)。反之,使用Trametinib药物抑制ERK活化可显著降低内源性Thr284磷酸化水平(图3n,o)。这些发现共同证明ERK特异性磷酸化VISTA的Thr284位点。
作者接下来研究ERK1介导的磷酸化如何影响VISTA-TRIM25相互作用。异位表达ERK1可提高pT284-VISTA水平并增强VISTA与TRIM25的相互作用(图3p)。相反,通过Trametinib抑制ERK1会降低pT284-VISTA水平并削弱VISTA与TRIM25的相互作用(图3q)。与这些发现一致,异位表达ERK1可增加TRIM25介导的VISTA K63连接型泛素化,而Trametinib处理则降低此修饰(图3r,s)。此外,VISTA T284A突变体与TRIM25的相互作用减弱,且K63连接型泛素化水平降低(图3t)。
在功能方面,异位表达ERK1和BRAF激酶可显著提高VISTA蛋白丰度,而作者检测的其他激酶无此效应(图3u)。相反,使用Trametinib抑制ERK活化则显著降低VISTA蛋白丰度(图3v-x)。而且,与野生型VISTA相比,T284A突变体形式的VISTA显示出更短的半衰期。这些结果共同表明,ERK通过保守的D结构域与VISTA相互作用并磷酸化其Thr284位点。该磷酸化被14-3-3ε识别,进而促进VISTA与TRIM25的结合,增强K63连接型泛素化并稳定VISTA蛋白。
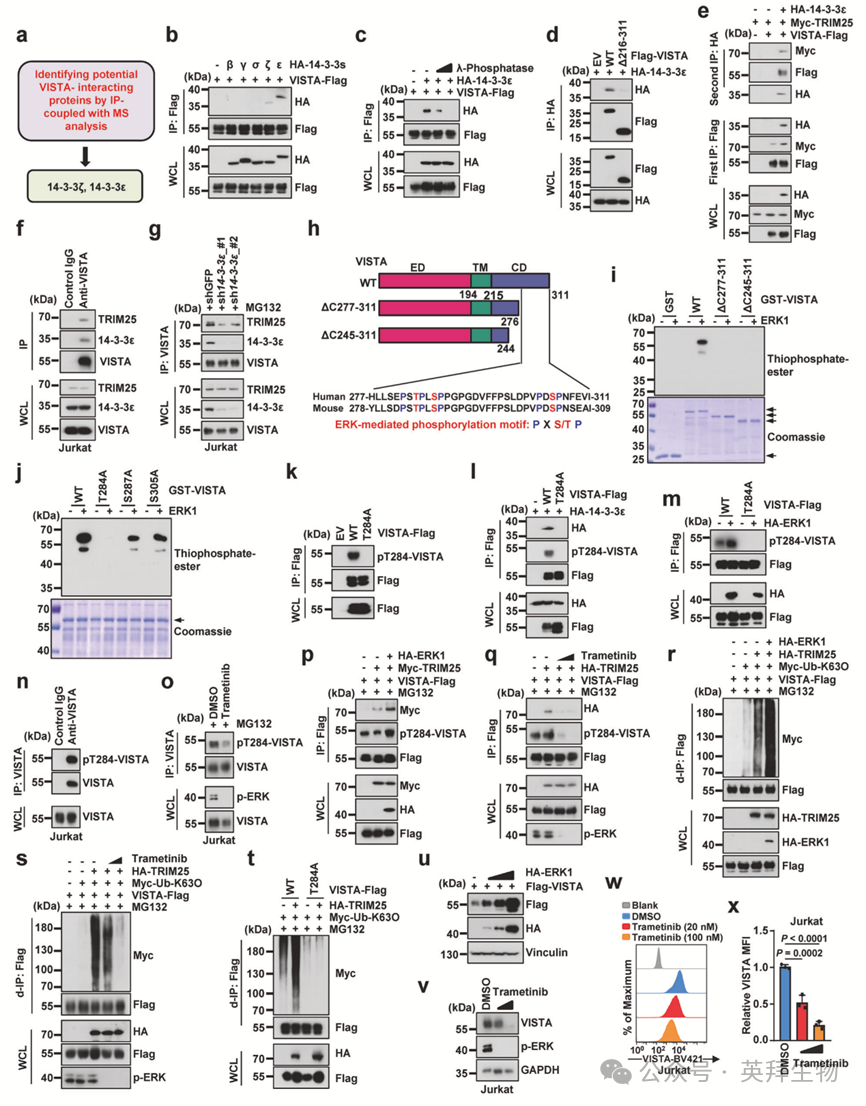
图3:14-3-3ε识别ERK介导的VISTA Thr284磷酸化,促进VISTA与TRIM25相互作用
4)T细胞条件性敲除Trim25的小鼠表现出正常表型和T细胞发育
与既往研究一致,Vista和Trim25在免疫器官(脾脏、淋巴结、胸腺)及具有显著白细胞浸润的肺组织中高表达(图4a,b)。鉴于这些免疫器官富含在抗肿瘤免疫应答中起关键作用的T淋巴细胞,作者构建了T细胞特异性敲除Trim25的小鼠,以探究Trim25在调控VISTA蛋白稳态和T细胞(特别是CD8+ T细胞)功能中的生理意义。为此,作者采用将Trim25 floxed等位基因小鼠与Cd4-Cre小鼠交配的育种策略,成功获得Trim25fl/flCd4-Cre(命名为Trim25 cKO)小鼠。作者证实从Trim25 cKO小鼠脾脏分离的CD8+ T细胞中Trim25蛋白被成功敲除。
如既往全身性Trim25敲除小鼠研究所示,Trim25 cKO小鼠也表现出正常的生长和体重特征,且符合预期的孟德尔遗传比例和性别比例。在稳态条件下,Trim25缺失对胸腺、脾脏和淋巴结中的T细胞发育未产生显著影响,因为Trim25 cKO小鼠来源的CD4+单阳性、CD8+单阳性和CD4+CD8+双阳性细胞比例与野生型小鼠相当。同样,在稳态条件下,6周龄野生型与Trim25 cKO小鼠脾脏中的初始、效应记忆和中央记忆CD4+及CD8+细胞亚群比例无显著差异。这些发现共同表明,T细胞中Trim25缺失在稳态条件下未明显影响T细胞发育或小鼠生长。
5)敲除Trim25可降低T细胞中VISTA表达并增强抗肿瘤免疫
经抗CD3/CD28刺激后,Trim25缺陷型T细胞中VISTA蛋白水平显著降低,而其他免疫检查点(包括PD-1、CTLA-4和TIM-3)的表达保持不变(图4c,d)。这进一步支持Trim25在T细胞中作为VISTA蛋白丰度正向调控因子的生理功能。值得注意的是,在相同刺激条件下,与野生型T细胞相比,Trim25敲除T细胞的活化标志物CD69表达增加,且细胞毒性因子干扰素-γ、肿瘤坏死因子及细胞毒性效应分子颗粒酶B的产量升高(图4e-g)。尽管存在这些差异,抗CD3/CD28刺激后野生型与Trim25缺陷型T细胞的凋亡水平相当。这些发现共同提示Trim25缺失可增强CD8+ T细胞的体外活化。
既往研究表明T细胞中VISTA缺失主要通过促进T细胞活化来改善抗肿瘤免疫应答。鉴于作者上述结果证明T细胞中敲除Trim25可降低VISTA蛋白水平并增强体外T细胞活化,作者探究了T细胞中Trim25缺失对小鼠抗肿瘤免疫应答的影响。为此,作者在野生型和Trim25 cKO小鼠皮下接种结直肠癌MC38细胞,并每两天监测肿瘤体积。结果显示,与野生型小鼠相比,Trim25 cKO小鼠皮下MC38肿瘤的生长显著延缓(图4h-j)。在同系小鼠Lewis肺癌肿瘤模型中也观察到类似结果,与野生型小鼠相比,Trim25 cKO小鼠肿瘤生长减缓且生存期延长。
为更深入理解T细胞中Trim25表达对肿瘤免疫微环境的影响,作者对从野生型或Trim25 cKO C57BL/6J小鼠采集的MC38移植瘤中分离的CD45+免疫细胞进行单细胞RNA测序。经过质控和过滤,作者共捕获33,990个细胞并获得其单细胞转录组数据,通过均匀流形近似与投影方法鉴定出20个转录同质细胞簇。基于转录特征和SingleR软件包,作者将这些细胞簇注释为11种细胞类型:CD4+ T细胞、CD8+ T细胞、自然杀伤细胞、巨噬细胞、单核细胞、B细胞、浆细胞样树突状细胞、经典树突状细胞1、经典树突状细胞2、中性粒细胞和肥大细胞。
聚焦CD8+ T细胞,作者通过无监督UMAP分析将3736个CD8+ T细胞分为初始CD8+ T细胞、早期活化T细胞、CD8+效应T细胞、耗竭CD8+ T细胞、组织驻留记忆CD8+ T细胞和CD8+周期T细胞(图4k)。与野生型小鼠相比,作者观察到Trim25 cKO小鼠来源的CD8+ Teff细胞群体增加(图4l)。此外,对肿瘤浸润Trim25 KO CD8+ T细胞中上调基因的转录组分析和基因本体富集分析显示,与T细胞活化、免疫效应过程及白细胞细胞间粘附相关的通路显著富集(图4m,n)。而且,与野生型相比,Trim25 KO CD8+ T细胞中涉及T细胞效应分子的基因如Ifng和Gzmb表达上调(图4o,p)。
随后,对肿瘤浸润免疫细胞的流式细胞术分析显示,与野生型小鼠相比,Trim25 cKO小鼠来源的MC38肿瘤中浸润CD8+ T细胞的VISTA蛋白水平显著降低(图4q)。虽然野生型与Trim25 cKO小鼠肿瘤浸润CD4+和CD8+ T细胞比例相似,但Trim25 cKO小鼠这些T细胞群体中关键细胞因子和效应分子的产生显著升高(图4r-t)。同时,野生型与Trim25 cKO小鼠肿瘤中的调节性T细胞、髓源性抑制细胞、巨噬细胞、树突状细胞比例无显著差异。这些结果共同表明,T细胞中Trim25缺失可能增强肿瘤微环境中CD8+ T细胞的活化及细胞毒性杀伤功能,从而改善对肿瘤生长的控制。
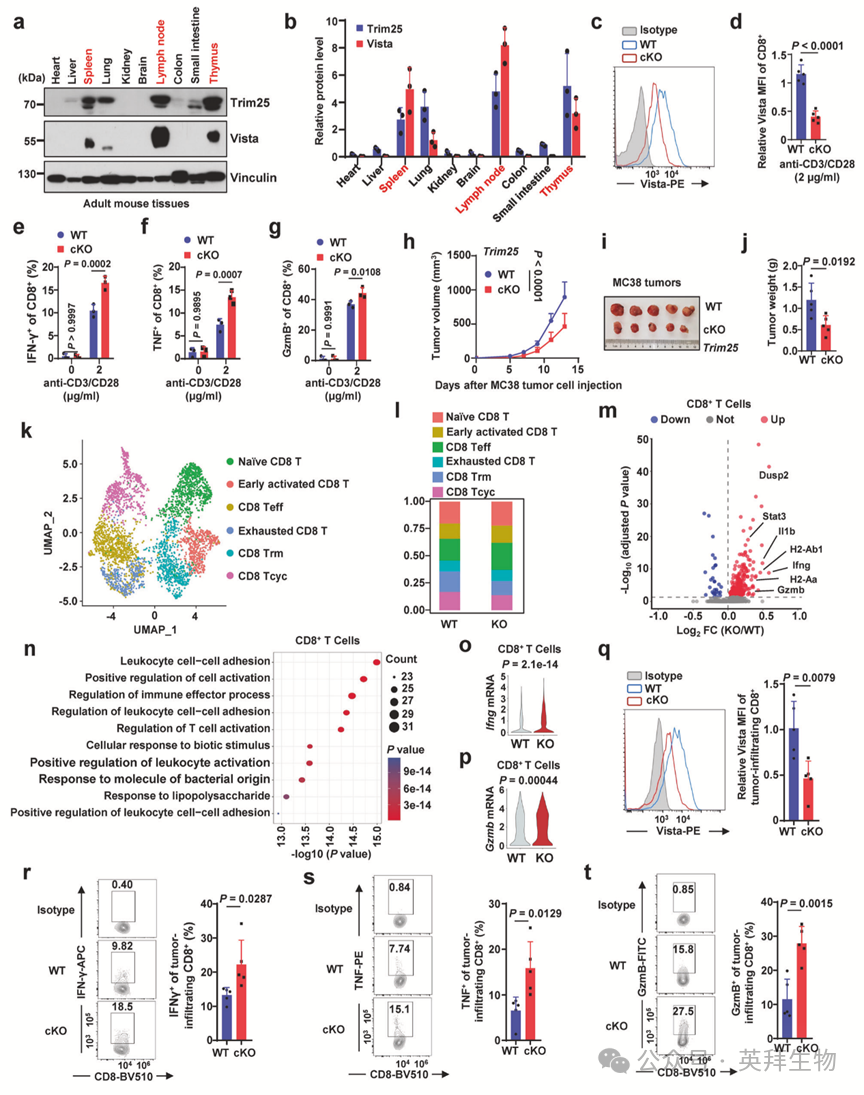
图4:Trim25敲除降低T细胞中VISTA表达并增强抗肿瘤免疫
6)在自体结直肠肿瘤模型中Trim25 cKO小鼠表现出更强的肿瘤控制能力
为进一步确定T细胞中Trim25缺失是否影响自体结直肠肿瘤发生,作者采用成熟的氧化偶氮甲烷/葡聚糖硫酸钠诱导的小鼠结直肠肿瘤模型。对野生型和Trim25 cKO小鼠进行AOM/DSS处理以诱导结直肠肿瘤发展(图5a)。在AOM/DSS处理前,野生型与Trim25 cKO小鼠的结直肠特征无显著差异。然而,经AOM/DSS处理后,Trim25 cKO小鼠的结直肠肿瘤数量较野生型对照显著减少(图5b-e)。值得注意的是,在Trim25 cKO小鼠来源的肿瘤中观察到肿瘤增殖减少及肿瘤细胞死亡增加(图5f,g)。如在皮下MC38小鼠模型中观察到的,野生型与Trim25 cKO小鼠在AOM/DSS诱导的结直肠肿瘤中CD4+或CD8+ T细胞比例无显著差异。然而,与野生型小鼠相比,Trim25 cKO小鼠AOM/DSS诱导的结直肠肿瘤中浸润T细胞的关键效应细胞因子产生显著增加(图5h-j)。此外,免疫组化分析显示,与野生型对照相比,Trim25 cKO小鼠AOM/DSS诱导肿瘤中Vista蛋白表达显著降低。这些结果共同表明Trim25缺失可能促进T细胞活化增强,从而产生更强烈的抗肿瘤免疫应答。
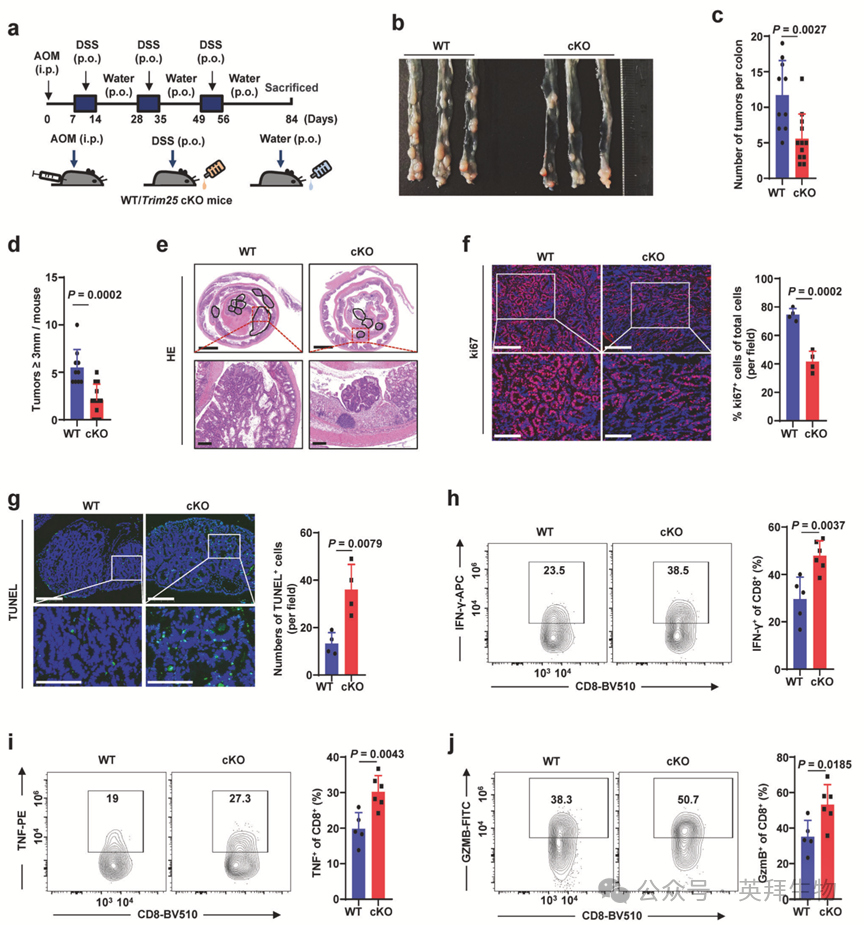
图5:Trim25条件性敲除小鼠在自体肿瘤模型中表现出更强的肿瘤控制能力
7)携带肿瘤的Trim25 cKO小鼠对抗PD-L1免疫治疗反应更佳
VISTA通路已被证实与黑色素瘤或前列腺癌患者分别接受抗PD-1或抗CTLA-4治疗时获得性耐药机制相关。此外,已知VISTA与PD-1/PD-L1在调控T细胞免疫应答中具有不同作用[30]。因此作者推测,导致T细胞中VISTA水平降低的Trim25缺失是否可能增强PD-1/PD-L1阻断疗法的疗效。为验证该假设,作者对携带皮下MC38肿瘤的野生型或Trim25 cKO小鼠分别给予对照IgG或抗PD-L1抗体治疗(图6a)。值得注意的是,与其他组别相比,接受抗PD-L1治疗的Trim25 cKO小鼠表现出最显著且有效的肿瘤控制效果(图6b-d)。此外,与其他组别相比,经抗PD-L1抗体治疗的Trim25 cKO小鼠来源肿瘤显示Vista水平降低,同时肿瘤浸润CD8+ T细胞中效应细胞因子的产生显著增加(图6e-g),提示抗PD-L1抗体治疗的Trim25 cKO小鼠免疫应答增强。在涉及抗PD-L1治疗的皮下MC38肿瘤模型中观察到的结果一致,在AOM/DSS诱导的结直肠癌模型中也获得类似发现。具体而言,在各实验组中,接受抗PD-L1治疗的Trim25 cKO小鼠对AOM/DSS诱导肿瘤生长的抑制效果最为显著(图6h-j)。
为验证Trim25缺失引起的肿瘤生长抑制是否主要源于VISTA蛋白减少,作者使用抗VISTA抗体处理携带移植MC38肿瘤的野生型和Trim25 cKO小鼠。在野生型小鼠中,抗VISTA抗体给药显著抑制肿瘤生长(图6k,l),而Trim25 cKO小鼠较野生型小鼠肿瘤生长减缓。然而,与IgG对照治疗相比,抗VISTA抗体治疗未在Trim25 cKO小鼠中进一步显著降低肿瘤生长(图6k,l)。免疫组化分析证实Trim25 cKO小鼠肿瘤中Vista蛋白水平较野生型小鼠显著降低。这些发现提示TRIM25缺失可重现VISTA免疫检查点阻断的效果,并可能作为通过降低T细胞中VISTA表达来抑制肿瘤生长的内在策略。
鉴于ERK介导的VISTA Thr284磷酸化对TRIM25依赖的VISTA蛋白稳定至关重要(图3),作者探究了ERK抑制能否增强免疫治疗效果。在携带MC38肿瘤的野生型和Trim25 cKO小鼠中,Trametinib联合抗PD-L1抗体治疗较单一治疗显著抑制肿瘤生长(图6m-o)。值得注意的是,在所有组别中,接受联合治疗的Trim25 cKO小鼠肿瘤生长抑制最为显著。与这些结果一致,经联合治疗的Trim25 cKO小鼠肿瘤浸润CD8+ T细胞中效应分子表达水平最高,提示靶向ERK1介导的VISTA磷酸化可增强T细胞介导的抗肿瘤免疫。
为进一步靶向TRIM25-VISTA相互作用,作者设计了包含Thr284位点的细胞穿透性VISTA肽段,分别包含未修饰Thr284或Thr284磷酸化形式,并与TAT序列融合。显著的是,与TAT-VISTA野生型肽段相比,TAT-VISTA pT284肽段处理显著抑制VISTA与TRIM25的相互作用,并降低细胞内VISTA表达(图6p,q)。此外,在携带MC38肿瘤的小鼠中,TAT-VISTA pT284肽段联合抗PD-L1抗体治疗较单药治疗显著抑制肿瘤生长(图6r-t)。免疫组化证实经TAT-VISTA pT284肽段治疗的小鼠肿瘤中VISTA表达降低。这些结果表明,使用TAT-VISTA pT284肽段靶向TRIM25-VISTA轴可能为抗肿瘤免疫提供潜在治疗策略。
为进一步验证TRIM25-VISTA轴在肿瘤细胞和免疫细胞中的双重作用,作者构建了稳定表达野生型VISTA或磷酸化缺陷突变体VISTA-T284A的MC38细胞(该细胞系内源性不表达VISTA),并将其皮下移植至野生型或Trim25 cKO小鼠。在野生型小鼠中,表达VISTA-T284A的肿瘤比表达VISTA-WT的肿瘤生长更缓慢。然而在Trim25 cKO小鼠中,表达VISTA-T284A的肿瘤相对于所有其他组别(包括野生型小鼠中的VISTA-WT和T284A肿瘤)均表现出显著生长受损。此外,与VISTA-WT对照相比,VISTA-T284A肿瘤细胞中VISTA蛋白表达显著降低。这些数据共同支持了一个模型:VISTA通过其在肿瘤细胞和T细胞中的表达促进免疫抑制。ERK激酶对Thr284的磷酸化促进了TRIM25介导的VISTA稳定,该通路可通过遗传或药理学手段破坏。因此,通过基因敲除、ERK抑制或肽段干预等方式双重靶向肿瘤细胞和免疫细胞中的TRIM25-VISTA轴,可能显著增强免疫检查点阻断疗法的疗效。
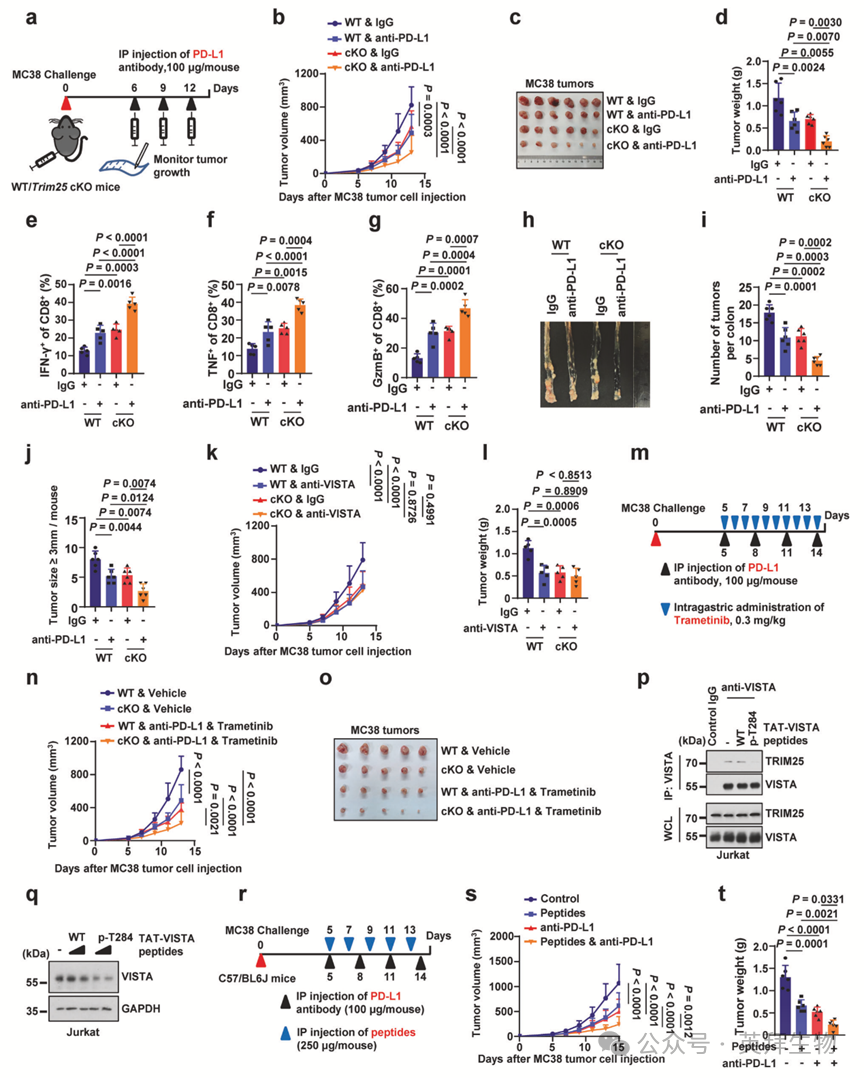
图6:T细胞中Trim25缺失增强免疫检查点阻断疗效
8)Trim25 KO CAR-T细胞展现增强的抗肿瘤疗效
鉴于Trim25缺失会导致VISTA水平降低和T细胞活化水平升高,作者研究了Trim25敲除是否可能增强CAR-T疗法的效果。为此,作者分别制备了野生型和Trim25 KO的抗CD19 CAR-T细胞。从脾脏分离的野生型或Trim25 KO CD3+ T细胞,用携带由hCD19单链片段和小鼠CD28及CD3ζ胞质结构域组成的CAR的慢病毒进行感染(称为抗CD19 CAR-T细胞)(图7a)。将工程化表达人CD19蛋白的MC38细胞皮下植入C57BL/6J小鼠。然后通过尾静脉静脉注射给予抗CD19 CAR-T细胞(图7b)。与野生型抗CD19 CAR-T细胞相比,Trim25 KO抗CD19 CAR-T细胞表现出增强的抗肿瘤反应,导致肿瘤生长被显著抑制(图7c-e)。值得注意的是,作者还观察到在肿瘤浸润的Trim25 KO CAR-T细胞中,Vista水平显著降低,而IFN-γ、TNF和GzmB的产生则显著增加(图7f-i)。
为进一步评估Trim25缺失是否通过降低Vista表达来增强CAR-T疗法的疗效,作者制备了表达小鼠Vista蛋白的Trim25 KO抗CD19 CAR-T细胞(图7j,k)。在用异位表达Vista的Trim25 KO抗CD19 CAR-T细胞治疗后,小鼠的MC38肿瘤生长速度快于Trim25 KO抗CD19 CAR-T细胞组(图7l)。这些发现共同表明,T细胞中敲除Trim25能显著增强CAR-T细胞疗法在小鼠肿瘤模型中的效果。
在确定K217是TRIM25介导的VISTA K63连接型泛素化的关键位点后,作者接下来评估了其功能相关性。为此,作者分别从野生型和Vista KO C57BL/6J小鼠分离T细胞,并制备了用野生型VISTA或K217R突变型VISTA重建的Vsir KO抗CD19 CAR-T细胞。然后将这些细胞用于荷有MC38-CD19肿瘤的C57BL/6J小鼠的过继性细胞转移实验。体内肿瘤生长实验表明,用野生型VISTA重建完全恢复了在Vsir KO CAR-T细胞中丧失的促肿瘤效应。相比之下,用缺乏K63连接型泛素化的K217R突变体重建仅部分挽救了该效应。接受VISTA-K217R CAR-T细胞的小鼠表现出更好的肿瘤控制效果。流式细胞术分析显示,与野生型对照相比,VISTA-K217R CAR-T细胞中VISTA表达部分降低。这些发现强调了TRIM25介导的K217位点K63连接型泛素化在稳定VISTA和促进免疫逃逸中的关键作用。
总之,T细胞中Trim25的缺失通过降低VISTA表达和提升T细胞效应器功能来增强抗肿瘤免疫。这些效应与免疫检查点阻断疗法产生协同作用,并显著改善小鼠肿瘤模型中的CAR-T细胞疗法。靶向TRIM25-VISTA轴,特别是通过干扰ERK1介导的VISTA磷酸化或泛素化,为克服当前癌症免疫疗法的耐药性提供了一个前景广阔的途径。
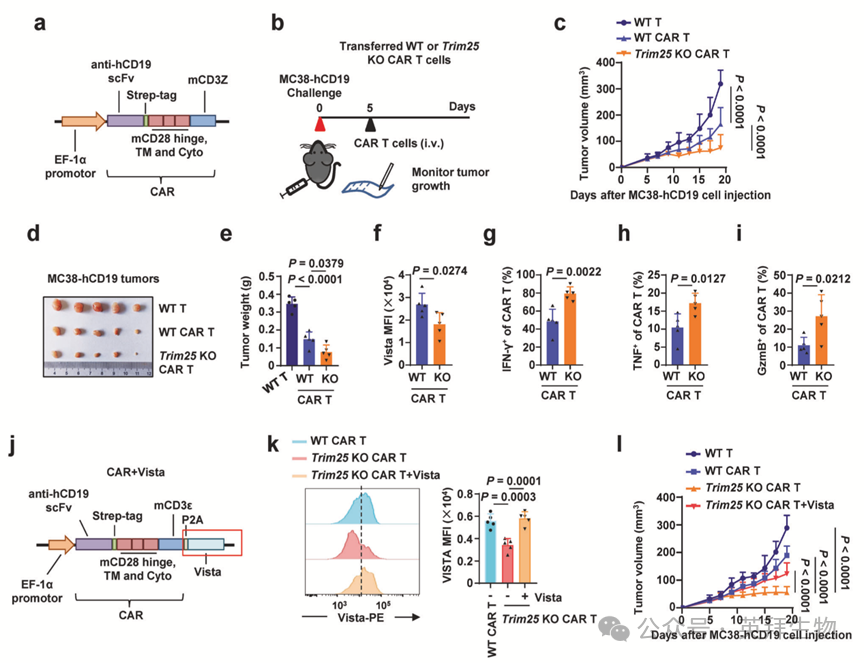
图7:Trim25敲除CAR-T细胞展现增强的抗肿瘤疗效
结论:
综上所述,作者首次揭示了TRIM25通过介导VISTA的K63连接型泛素化并拮抗其K48连接型降解途径来稳定VISTA蛋白的分子机制,同时发现ERK激酶通过磷酸化VISTA第284位苏氨酸增强其与TRIM25的相互作用。该研究不仅阐明了VISTA蛋白稳态调控新通路,更通过基因敲除和肽段干预实验证明靶向TRIM25-VISTA轴能够显著增强免疫检查点阻断疗法和CAR-T细胞的抗肿瘤疗效,为克服肿瘤免疫治疗耐药性提供了新的策略和靶点。
参考文献:
Sun Y, Zhang Z, Li H, Bu X, Chen L, Wang X, Fan L, Chen B, Kong L, Dai P, Song W, Xiao X, Shi J, Xiang B, He C, Yao Y, Xiong W, Yu H, Jiang C, Qian Q, Liu H, Tian S, Qing G, Yang Z, Wei W, Freeman GJ, Zhu H, Zhang J. Destruction of VISTA by TRIM25 ablation in T cells potentiates cancer immunotherapy. Cell Res. 2025 Nov 13. doi: 10.1038/s41422-025-01186-5IF: 25.9 Q1 . Epub ahead of print. PMID: 41225152.