






AARS1介导的H3K18与STAT1乳酸化修饰促进糖尿病肾病中铁死亡的发生
糖尿病肾病(DN)是全球范围内导致终末期肾病的主要原因。近期研究揭示,乳酸介导的组蛋白乳酸化作为一种新型表观遗传修饰,参与了糖尿病相关并发症的发生与发展。然而,乳酸转移酶在DN中的作用尚不明确。丙氨酰-tRNA合成酶1(AARS1)被鉴定为一种新型乳酸转移酶,可调控组蛋白H3第18位赖氨酸的乳酸化修饰(H3K18la)。本研究旨在探究AARS1介导的H3K18la是否参与DN的发病机制,并进一步阐明其潜在的作用机理。本研究采用野生型与丙氨酰-tRNA合成酶1(AARS1)杂合子(AARS1+/–)小鼠构建的DN模型。通过转录组学与脂质组学分析,并结合多种分子生物学方法,系统阐明了AARS1调控DN中铁死亡过程的潜在机制。研究结果表明,在DN模型中,AARS1与H3K18la的表达升高通过调控铁死亡参与肾功能障碍和肾细胞死亡进程。此外,AARS1通过增加脂肪酸延长酶-5(ELOVL5)的转录诱导脂质过氧化,最终导致铁死亡的发生。进一步研究发现,AARS1与信号转导和转录激活因子1(STAT1)相互作用,共同调控ELOVL5的转录;而使用STAT1特异性抑制剂氟达拉滨可延缓DN进展。同时,研究观察到AARS1通过调控STAT1和H3K18的乳酸化修饰来调节ELOVL5转录,从而触发铁死亡。通过β-丙氨酸抑制AARS1诱导的乳酸化可减轻DN模型小鼠和高糖状态细胞的铁死亡程度。本研究证实,在糖尿病肾病模型中,AARS1通过诱导H3K18和STAT1的乳酸化修饰调控ELOVL5转录,进而触发铁死亡过程。该研究于2025年10月发表在《Cell Death & Differentiation》,IF:15.4。
技术路线:
主要研究结果:
1.AARS1与H3K18la在糖尿病肾病(DN)患者及模型中的表达上调
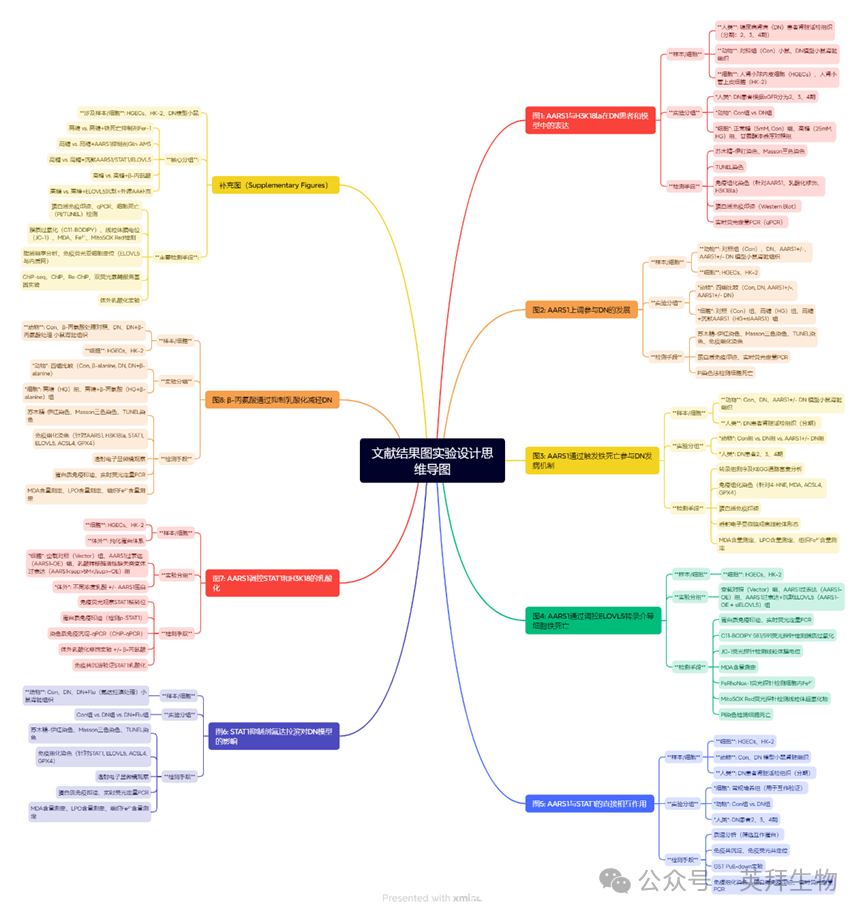
本研究纳入的DN患者临床特征见补充表1。根据估算肾小球滤过率(eGFR),DN患者被分为2期、3期或4期。图1A展示了DN患者肾脏样本的苏木精-伊红(HE)染色结果。Masson三色染色显示,随着DN分期的进展,胶原沉积与间质纤维化程度显著加重(图1A)。与此一致的是,TUNEL检测结果表明,随着DN分期进展,患者肾脏中的细胞死亡数量也相应增加(图1A)。此外,细胞死亡现象在肾小管和肾小球中均被观察到(图1A)。既往研究[21-23]提示H3K18la可能参与糖尿病相关并发症的发生与发展。同时,已有研究证实AARS1是调控H3K18la的新型乳酸转移酶[24,25]。因此,我们对DN患者肾脏组织中AARS1、乳酸化修饰及H3K18la的水平进行了检测。数据显示,随着DN分期的进展,患者肾脏中AARS1、乳酸化修饰及H3K18la的水平均显著升高(图1A)。本研究中小鼠的血清生化指标见补充表2。苏木精-伊红(HE)和Masson三色染色显示,DN小鼠肾脏发生显著病理改变(图1B)。TUNEL染色结果表明,DN小鼠肾脏的细胞死亡数量有所增加(图1B)。此外,DN小鼠肾脏中AARS1蛋白、乳酸化修饰及H3K18la的水平均出现升高(图1B、C)。与此一致的是,AARS1的mRNA水平在DN小鼠肾脏中也呈现上调趋势(图1D)。
此外,高糖处理可使人肾小球内皮细胞(HGECs)和人肾小管上皮细胞(HK-2)中AARS1、乳酸化修饰及H3K18la的水平增加(补充图1A、B)。同时,高糖状态下的细胞死亡程度也显著增强(补充图1C)。值得注意的是,作为渗透压对照的甘露醇处理,对高糖细胞中的AARS1、乳酸化修饰、H3K18la水平以及细胞死亡均未产生影响(补充图1A–C)。以上数据共同表明,AARS1、乳酸化修饰及H3K18la在DN患者和疾病模型中均呈现表达上调。
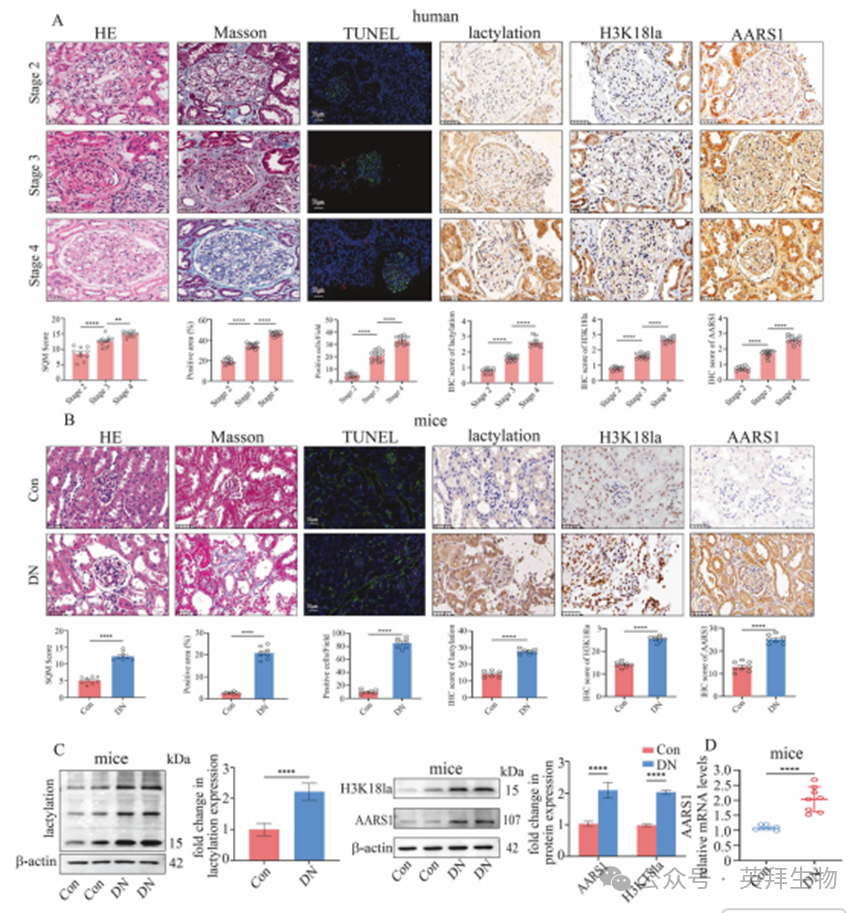
图1. AARS1与H3K18la在糖尿病肾病(DN)患者及模型中的表达上调
2.AARS1表达上调参与糖尿病肾病(DN)的疾病进程
为深入探究AARS1在DN中的作用,我们利用AARS1杂合子(AARS1+/–)小鼠构建了DN模型(AARS1+/– DN小鼠),相关实验设计见图2A–C。与DN小鼠相比,AARS1+/– DN小鼠表现出以下特征:肾脏组织结构损伤减轻、纤维化程度缓解、肾细胞死亡减少、AARS1蛋白水平及其介导的乳酸化修饰与H3K18la水平降低(图2A–C),同时肾功能指标得到改善(补充表2)。
与此相一致的是,在高糖细胞中沉默AARS1可降低AARS1及H3K18la的表达水平(图2D、E)。此外,沉默AARS1能够逆转高糖诱导的人肾小球内皮细胞(HGECs)和人肾小管上皮细胞(HK-2)的死亡(图2F)。进一步研究发现,AARS1抑制剂Gln-AMS能以浓度和时间依赖性的方式抑制AARS1及H3K18la的表达(补充图1D、E),并且该抑制剂处理也能减少高糖状态下的细胞死亡(补充图1F)。以上数据表明,AARS1诱导的H3K18la修饰参与了DN的疾病发展过程。
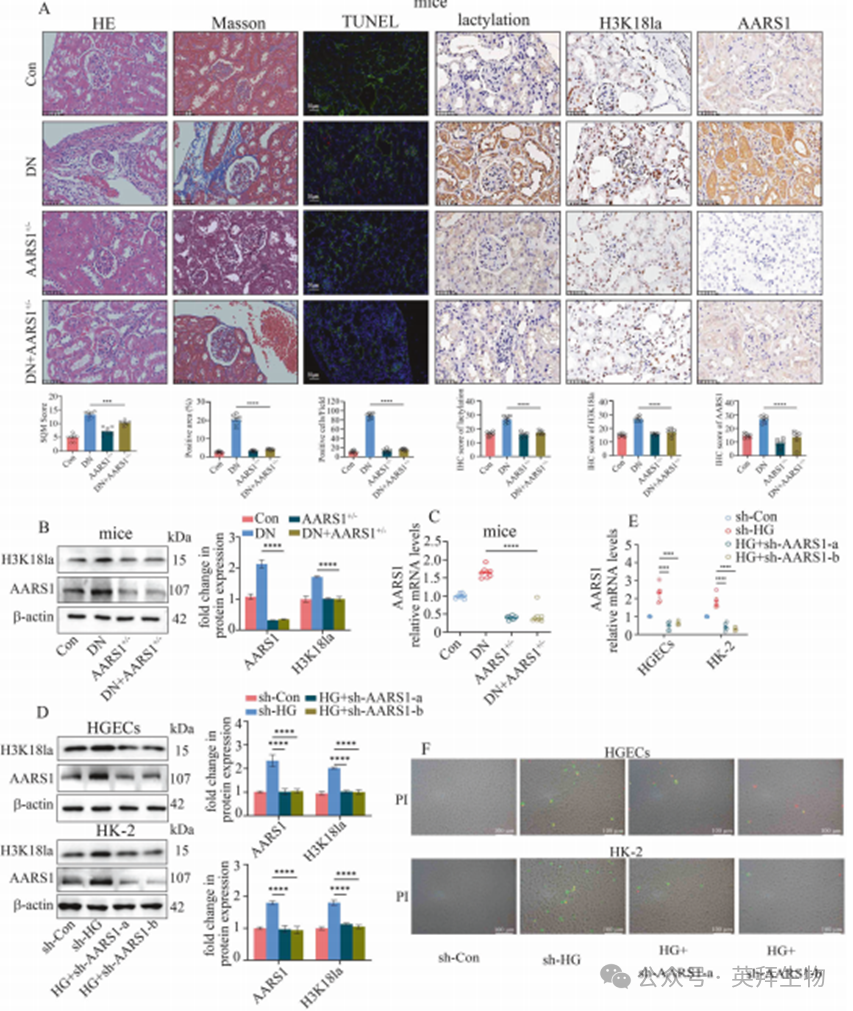
图2.AARS1表达上调参与糖尿病肾病(DN)的疾病进程
3.AARS1上调通过触发铁死亡参与糖尿病肾病(DN)的发病机制
为深入探究AARS1诱导的H3K18la修饰参与DN发病的机制,我们对小鼠肾脏组织进行了RNA-seq分析。数据显示,在DN小鼠肾脏中,与不饱和脂肪酸生物合成、脂肪酸延长、脂肪酸代谢及代谢通路相关的KEGG通路显著富集(图3A,补充表3)。既往研究提示,多不饱和脂肪酸(PUFA)生物合成通路在铁死亡过程中起关键作用[15]。与此相一致的是,随着DN分期进展,患者肾脏组织中4-羟基-2-壬烯醛(4-HNE)、丙二醛(MDA)及长链脂酰辅酶A合成酶4(ACSL4)水平逐渐升高,而谷胱甘肽过氧化物酶4(GPX4)水平逐渐降低(图3B),表明铁死亡参与了DN的发展过程。
我们进一步通过铁死亡抑制剂Ferrostatin-1(Fer-1)处理DN小鼠及高糖暴露细胞,验证铁死亡是否参与DN的进展。Fer-1处理可减轻肾脏组织损伤、抑制ACSL4表达并提高GPX4水平(补充图2A、B)。透射电子显微镜(TEM)观察显示,DN小鼠肾脏细胞呈现出线粒体结构致密、嵴显著减少、体积缩小等铁死亡特征性改变,而Fer-1处理能有效缓解这些超微结构异常(补充图2A)。此外,Fer-1还能降低DN小鼠肾脏MDA和脂质过氧化物(LPO)沉积、减少Fe²⁺含量并改善肾功能障碍(补充图2C–E;补充表2)。
在高糖细胞中,Fer-1能以时间和浓度依赖性的方式减少细胞死亡(补充图3A),同时降低ACSL4表达并提升GPX4水平(补充图3B)。采用脂质过氧化探针C11-BODIPY 581/591进行荧光染色,结果显示Fer-1可抑制高糖介导的脂质过氧化反应(补充图3C)。通过线粒体膜电位(MMP)探针JC-1的荧光染色发现,Fer-1处理能改善高糖诱导的HGECs和HK-2细胞MMP破坏(补充图3D),并降低高糖引起的MDA水平升高(补充图3E)。特异性Fe²⁺探针FeRhoNox-1荧光染色表明,Fer-1处理可降低高糖细胞内的Fe²⁺含量(补充图3F)。线粒体特异性超氧化物指示剂MitoSOX Red荧光染色显示,Fer-1处理能减少线粒体超氧化物的积累(补充图3G)。我们检测了DN模型中的铁死亡相关指标,以进一步证实AARS1诱导的H3K18la调控DN中铁死亡的过程。数据显示,与DN小鼠相比,AARS1+/– DN小鼠的肾脏表现出以下变化:ACSL4表达降低、GPX4水平升高、透射电镜(TEM)图像显示铁死亡形态改善、MDA与LPO含量减少、Fe²⁺含量下降(图3C–G)。
同样地,高糖处理的HGECs和HK-2细胞中铁死亡标志物水平升高,而这一现象可通过沉默AARS1得以逆转(补充图4)。与此一致的是,Gln-AMS处理能够抑制高糖细胞中的铁死亡过程(补充图5)。这些数据表明,AARS1诱导的H3K18la表达通过触发铁死亡参与DN的发病机制。
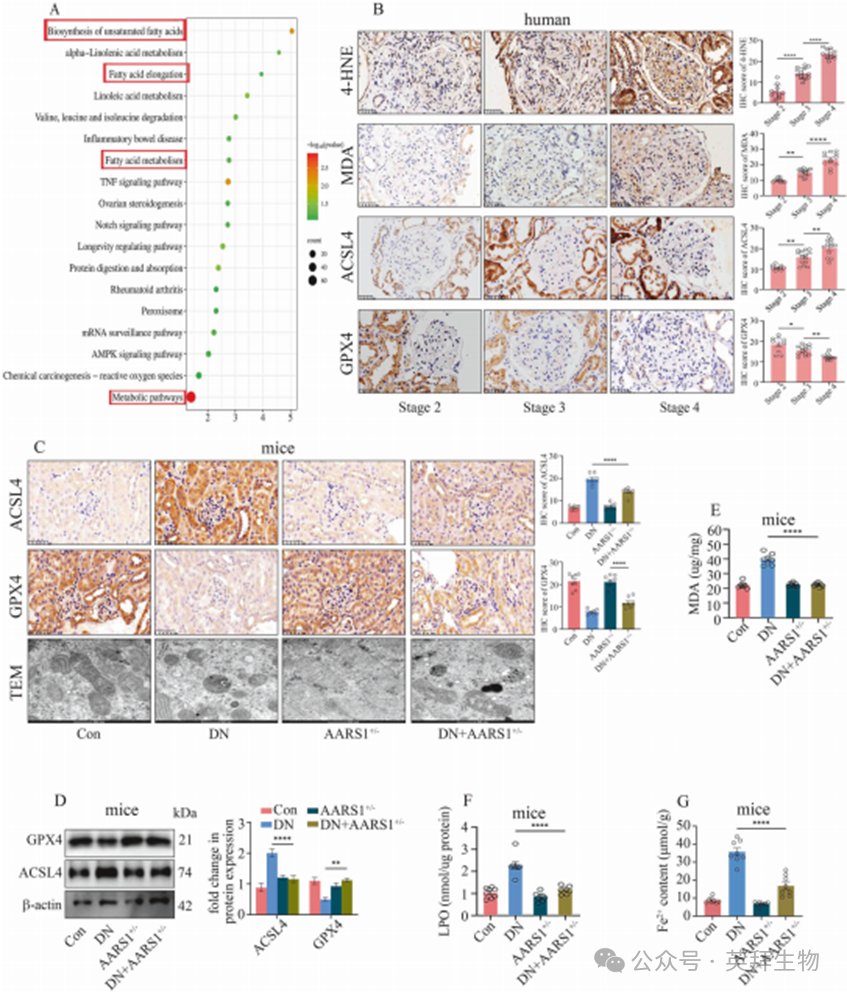
图3.AARS1上调通过触发铁死亡参与糖尿病肾病(DN)的发病机制
4.AARS1诱导的H3K18la通过调控ELOVL5转录参与糖尿病肾病(DN)模型中的铁死亡过程
接下来,我们深入探究了AARS1诱导的H3K18la参与DN患者铁死亡的具体机制。RNA-seq分析显示,脂肪酸延长酶5(ELOVL5)、反式-2,3-烯酰辅酶A还原酶(TECR)和3-羟基脂酰辅酶A脱水酶1(HACD1) 共同参与了不饱和脂肪酸生物合成、脂肪酸代谢、脂肪酸延长及代谢通路(补充图6A,补充表3)。值得注意的是,在这三个基因中,仅ELOVL5 出现在H3K18la的ChIP-seq结果中(补充表4)。ChIP实验证实,AARS1与H3K18la仅通过引物3(P3)富集在ELOVL5启动子区域;Re-ChIP实验进一步表明,AARS1与H3K18la共同占据ELOVL5启动子的相同区域(补充图6B)。
随着DN分期进展,患者肾脏中ELOVL5表达水平逐渐升高(补充图6C)。与之相一致的是,ELOVL5在DN模型小鼠(补充图6D–F)和高糖细胞(补充图6G、H)中均呈现上调表达。抑制ELOVL5表达可减轻高糖诱导的HGECs和HK-2细胞的铁死亡(补充图7)。对高糖处理的ELOVL5沉默细胞进行脂质代谢分析显示,包括花生四烯酸(AA)在内的多不饱和脂肪酸(PUFA)生成减少(补充图8A,补充表5)。免疫荧光数据显示,ELOVL5主要定位于内质网(补充图8B)。更重要的是,在ELOVL5沉默细胞中加入AA可逆转si-ELOVL5对高糖介导的铁死亡的保护作用(补充图8C–H)。此外,ELOVL5沉默也能减轻铁死亡诱导剂FINO2介导的铁死亡(补充图9)。这些数据表明,在DN患者中,ELOVL5通过调控内质网中的PUFA合成来触发铁死亡。
随后,我们验证了AARS1是否通过调控ELOVL5转录来介导DN患者的铁死亡。数据显示,过表达AARS1可增加细胞内ELOVL5表达(图4A、B)并促进铁死亡(图4)。而沉默ELOVL5则能消除AARS1过表达对铁死亡的促进作用(图4)。此外,抑制AARS1表达可降低DN模型小鼠肾脏(补充图10A–C)及高糖细胞(补充图10D–G)中的ELOVL5水平。这些数据共同表明,AARS1在DN模型中通过增强ELOVL5转录来促进铁死亡。
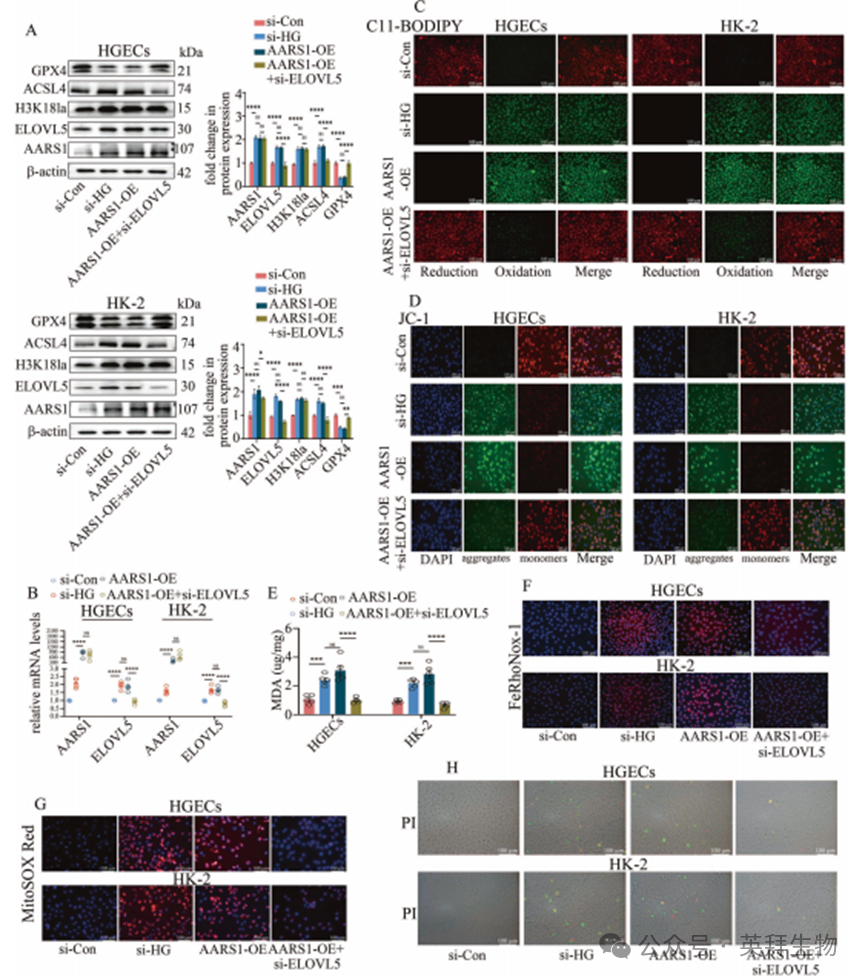
图4.AARS1诱导的H3K18la通过调控ELOVL5转录参与糖尿病肾病(DN)模型中的铁死亡过程
5.AARS1在体内外与信号转导和转录激活因子1(STAT1)直接相互作用
基因启动子区域的组蛋白修饰常与转录因子结合并存,共同调控下游基因的转录。为鉴定与AARS1相互作用的转录因子,本研究进行了质谱分析。数据显示,STAT1可能与AARS1存在相互作用(图5A,补充表6)。通过免疫共沉淀(Co-IP)和免疫荧光(IF)实验,我们证实STAT1在HGECs和HK-2细胞中与AARS1结合(图5B、C),而GST pull-down实验进一步验证了AARS1与STAT1之间的直接结合(图5D)。
此外,我们评估了STAT1在DN患者肾活检样本和DN模型中的表达情况。数据显示,STAT1水平随DN分期进展逐渐升高(图5E)。与此一致的是,STAT1在DN模型小鼠肾脏(图5F–H)及高糖细胞(图5I、J)中表达均呈现上调。
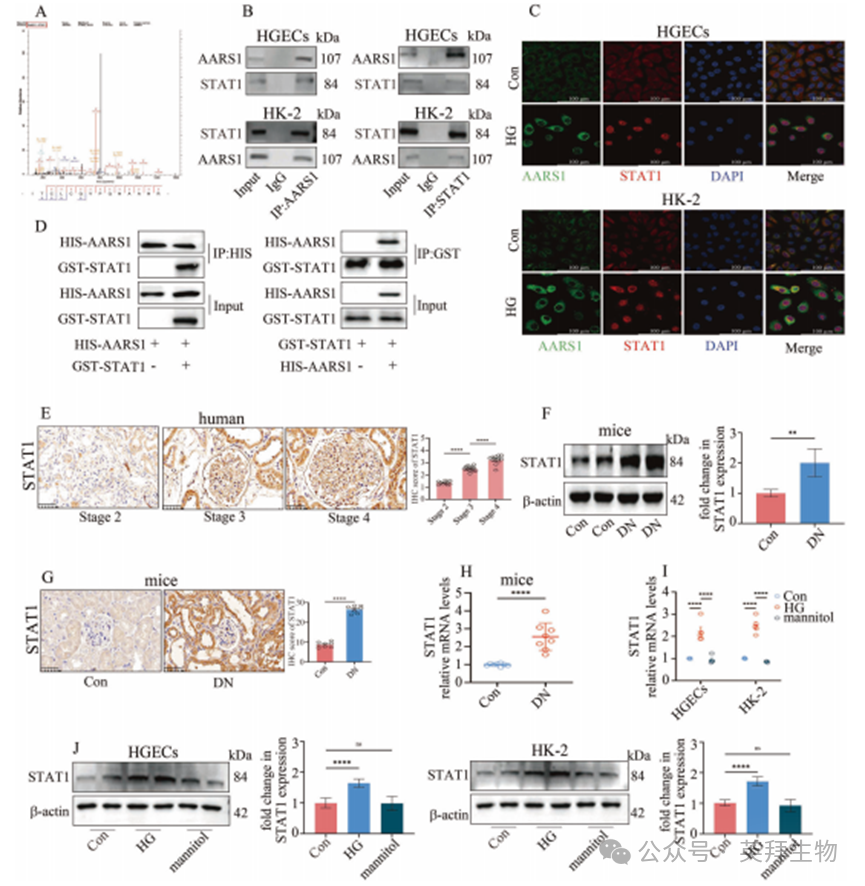
图5. AARS1在体内外与信号转导和转录激活因子1(STAT1)直接相互作用
6.STAT1通过调控ELOVL5转录参与糖尿病肾病(DN)模型中的铁死亡过程
为探究STAT1对ELOVL5表达及铁死亡的影响,我们进行了功能缺失与功能获得实验。沉默STAT1可降低ELOVL5水平,并抑制高糖诱导的HGECs和HK-2细胞铁死亡(补充图11)。相反,过表达STAT1可上调ELOVL5表达并激活铁死亡,而这一效应可被沉默ELOVL5所消除(补充图12)。为验证STAT1是否直接调控ELOVL5转录,本研究进行了染色质免疫沉淀(ChIP)和荧光素酶报告基因实验。ChIP实验显示STAT1富集于ELOVL5启动子区域,Re-ChIP实验进一步证实STAT1与AARS1共同占据ELOVL5启动子的相同区域(补充图13A)。通过JASPAR数据库(https://jaspar.genereg.net/)预测STAT1在ELOVL5启动子区域的潜在结合位点(见补充表7),其基序标识和位置权重矩阵分别展示于上下两栏(补充图13B)。随后,我们构建了这两个结合位点突变的质粒,发现突变后ELOVL5转录活性显著受抑制(补充图13C)。因此,我们认为STAT1直接结合ELOVL5启动子区域并参与其转录调控。
为探究STAT1是否可作为抑制DN铁死亡的潜在靶点,我们进行了以下研究:在细胞实验中,使用STAT1特异性抑制剂氟达拉滨(Flu)处理。结果显示,氟达拉滨能以浓度和时间依赖性方式抑制STAT1表达(补充图14A)。此外,Flu不仅能抑制STAT1表达,还可降低ELOVL5水平(补充图14B、C),进而抑制下游铁死亡过程(补充图14B–I)。与此一致的是,在DN模型小鼠腹腔注射氟达拉滨可有效下调STAT1和ELOVL5表达(图6A–C),抑制铁死亡(图6A–F),减轻肾组织损伤(图6A),并显著改善肾功能障碍(补充表2)。这些数据表明,STAT1抑制剂在防治DN方面具有重要的临床转化潜力。
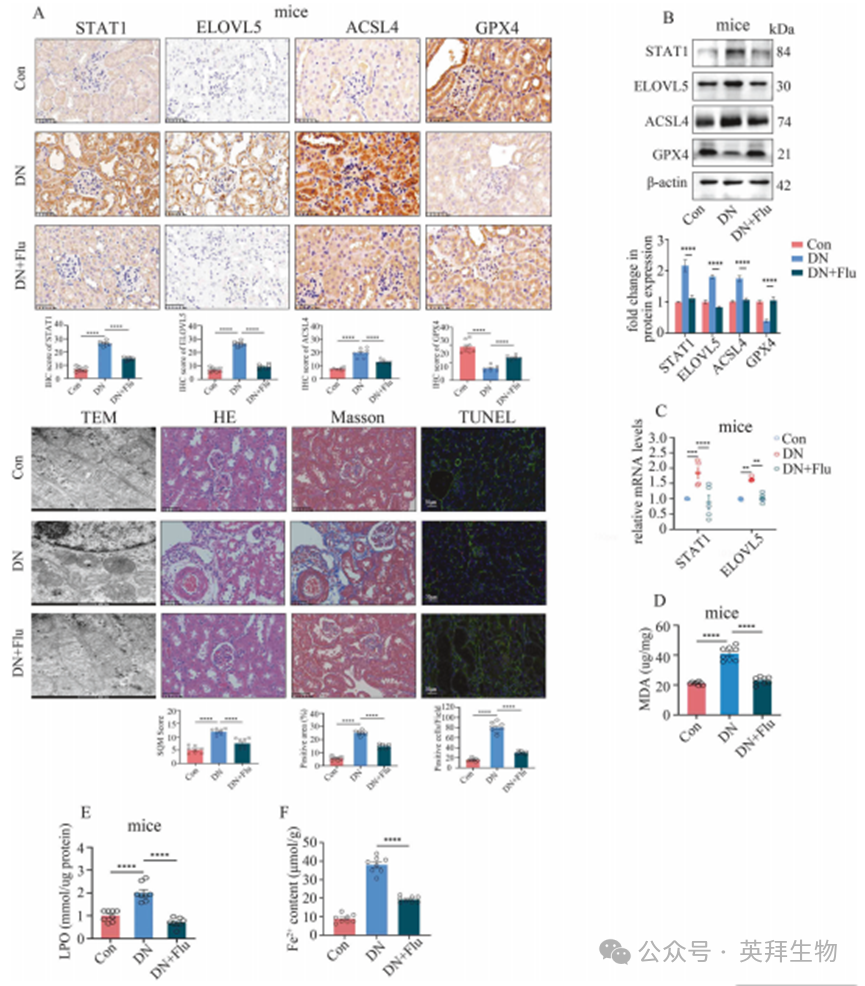
图6.STAT1通过调控ELOVL5转录参与糖尿病肾病(DN)模型中的铁死亡过程
7.AARS1通过调控STAT1和H3K18的乳酸化修饰调节ELOVL5转录并触发铁死亡
接下来,我们探究了AARS1是否影响STAT1的转录活性。数据显示,过表达AARS1可促进STAT1核转位(图7A)、增加STAT1磷酸化水平(图7B)并增强STAT1与ELOVL5启动子区域的结合(图7C)。然而,缺乏乳酸转移酶活性的AARS15M突变体[24]则无上述效应(图7A–C)。这些结果表明,AARS1可能通过调控STAT1的乳酸化修饰来增强其转录活性。
为验证这一假设,本研究采用了体内外乳酸化修饰实验。体外乳酸化实验显示,AARS1能以浓度依赖性方式利用乳酸诱导STAT1发生乳酸化修饰(图7D)。在HGECs和HK-2细胞中也证实AARS1可诱导STAT1乳酸化(图7E)。此外,过表达AARS1能上调STAT1、H3K18la及ELOVL5的表达(图7E),并促进细胞铁死亡(补充图15),而AARS15M则无此作用(图7E;补充图15)。
为阐明AARS1介导的STAT1乳酸化修饰对其转录功能的关键作用,我们根据文献报道[26]对STAT1的乳酸化位点进行了突变分析。数据显示,STAT1K193位点是受AARS1调控的最重要乳酸化位点(补充图16A)。更重要的是,突变STAT1K193位点可逆转AARS1过表达所诱导的STAT1核转位促进、磷酸化水平增加及与ELOVL5启动子结合增强等效应(补充图16B–D)。这些发现提示,在高糖环境中,AARS1通过催化STAT1第193位赖氨酸(K193)发生乳酸化修饰,从而增强STAT1的转录活性。综上所述,本研究证实AARS1通过调控STAT1与H3K18的双重乳酸化修饰,共同调节ELOVL5转录,进而触发DN中的铁死亡过程。
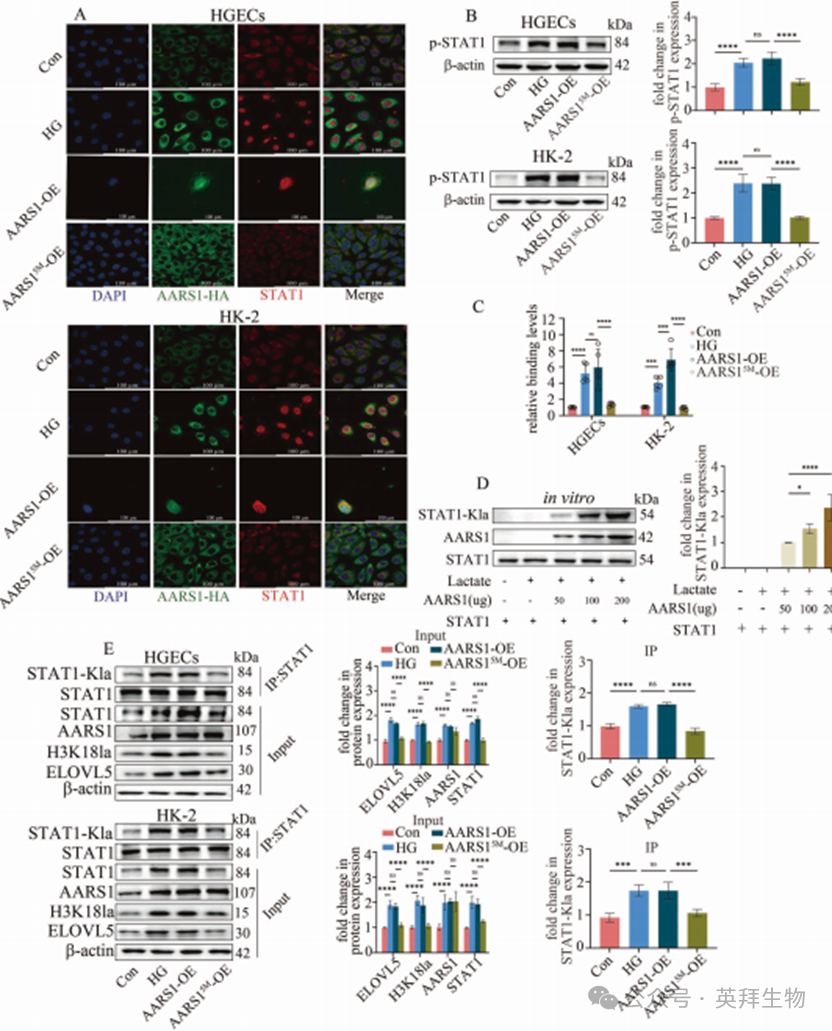
图7.AARS1通过调控STAT1和H3K18的乳酸化修饰调节ELOVL5转录并触发铁死亡
8.β-丙氨酸通过抑制AARS1诱导的乳酸化减轻糖尿病肾病模型小鼠及高糖细胞的铁死亡
鉴于AARS1介导的乳酸化修饰在糖尿病肾病(DN)中发挥重要作用,抑制该修饰过程可能成为DN的潜在治疗靶点。前期研究表明,β-丙氨酸可通过直接与乳酸竞争结合AARS1,有效拮抗AARS1诱导的乳酸化修饰[25]。我们的体外乳酸化实验证实,β-丙氨酸确实能拮抗AARS1诱导的H3K18和STAT1乳酸化(补充图17A、B)。随后,本研究采用β-丙氨酸处理DN模型。数据显示,β-丙氨酸能以浓度和时间依赖性的方式抑制高糖诱导的高糖细胞中STAT1和H3K18乳酸化(补充图17C)。此外,β-丙氨酸处理可降低高糖细胞中AARS1、STAT1、H3K18la及ELOVL5的表达,并抑制铁死亡过程(补充图18)。与此一致的是,在DN模型小鼠中,β-丙氨酸处理能下调AARS1、STAT1、H3K18la和ELOVL5的表达(图8A–C),抑制铁死亡(图8A–F),减轻肾组织损伤(图8A),并改善肾功能障碍(补充表2)。这些数据表明,通过β-丙氨酸抑制AARS1诱导的乳酸化修饰可能成为DN的有效治疗策略。
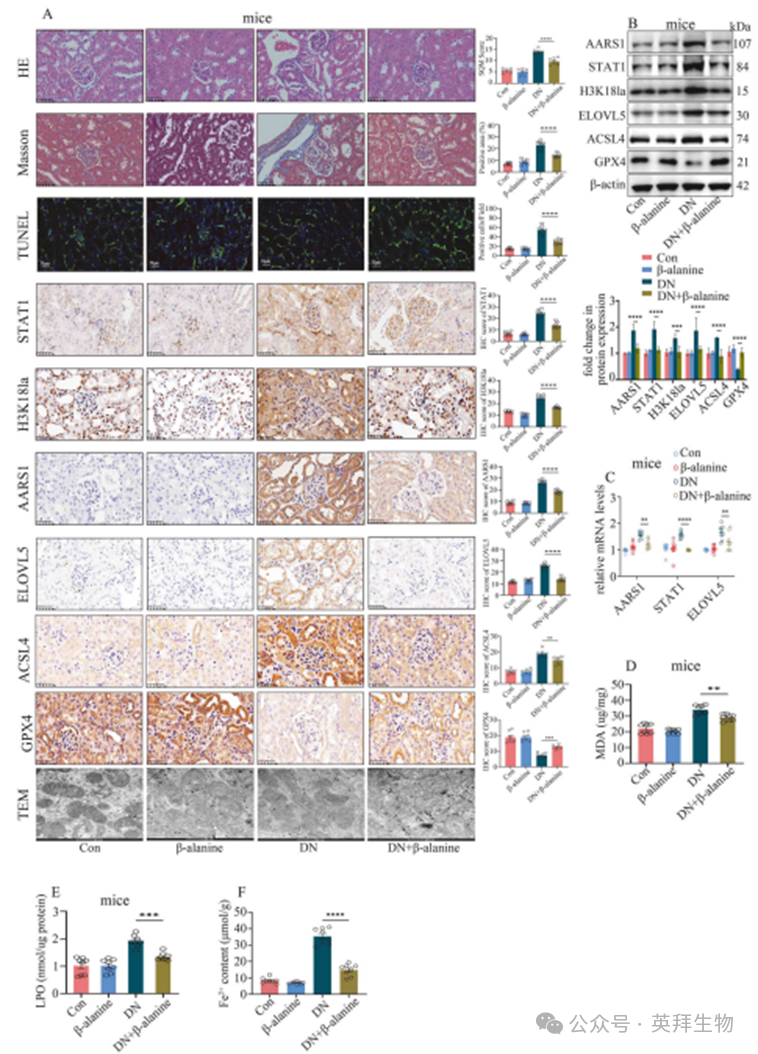
图8.β-丙氨酸通过抑制AARS1诱导的乳酸化减轻糖尿病肾病模型小鼠及高糖细胞的铁死亡
结论:
本研究揭示,AARS1通过催化组蛋白H3第18位赖氨酸(H3K18)和转录因子STAT1的乳酸化修饰,协同激活脂肪酸延长酶5(ELOVL5)的转录,进而促进多不饱和脂肪酸合成及脂质过氧化,最终触发铁死亡,驱动糖尿病肾病(DN)的发生与发展。抑制AARS1介导的乳酸化(如使用β-丙氨酸)或阻断STAT1活性(如使用氟达拉滨)均可有效减轻肾组织铁死亡与损伤,为DN提供了新的治疗靶点与策略。
参考文献:
Hong J, Xu H, Yu L, Yu Z, Chen X, Meng Z, Zhu J, Li J, Zhu M. AARS1-mediated lactylation of H3K18 and STAT1 promotes ferroptosis in diabetic nephropathy. Cell Death Differ. 2025 Sep 23.