






心脏发育新机制:GPAT4蛋白缺陷会引发“内部警报”,干扰心脏正常成型
心内膜在调控心肌发育中起着关键作用,理解其内在调控机制有助于阐明病理性心肌病的发生机制。甘油-3-磷酸酰基转移酶4(GPAT4)是一种锚定于内质网(ER)膜的蛋白质。尽管GPAT4在甘油磷脂生物合成中的作用已得到充分证实,但其在内质网中的功能尚不明确。本研究通过创建Gpat4全球性及组织特异性敲除小鼠模型,揭示GPAT4在心内膜发育中的关键作用。GPAT4缺失会诱发心内膜内质网应激反应,增强内质网-线粒体(ER-mito)通信,导致线粒体DNA(mtDNA)外溢。由此激活cGAS-STING通路,刺激I型干扰素反应,进而影响心脏发育。最终,通过抑制cGAS-STING-I型干扰素通路可挽救Gpat4敲除小鼠的心脏缺陷。这些发现揭示了GPAT4在维持心内膜及心脏发育过程中内质网稳态的关键作用,同时强调了cGAS-STING通路在心脏器官发生中的重要性。该研究于2025年4月发表在《Nature Communications》,IF:15.7。
技术路线
主要研究结果:
1.Gpat4基因全球敲除小鼠心脏发育缺陷
为探究GPAT4的生理功能,我们通过CRISPR-Cas9技术构建了Gpat4全基因敲除(KO)小鼠(图S1A)。结果发现Gpat4 KO小鼠在胚胎发育约E18.5阶段即出现胚胎致死现象(图S1B)。对胚胎发育第14.5天(E14.5)和第15.5天(E15.5)胚胎的解剖显示,Gpat4 KO小鼠存在明显的皮下水肿,提示心血管发育受损(图1A)。心脏形态学与组织学检查显示,Gpat4 KO小鼠心脏存在心室扩张、心肌壁变薄(Myo)及心肌小梁过度增生(Tra)(图1B–E、S1C)。KO心脏细胞增殖显著减少(图1F、G)。Western blot分析证实KO小鼠缺失GPAT4蛋白,其心脏组织中促凋亡蛋白BAX显著上调,而抗凋亡蛋白BCL2则大幅下调(图1H,I)。整体免疫荧光染色显示KO小鼠冠状血管形成严重受阻(图1J–N)。综合来看,这些发现揭示了GPAT4在胚胎心脏发育中的关键作用。
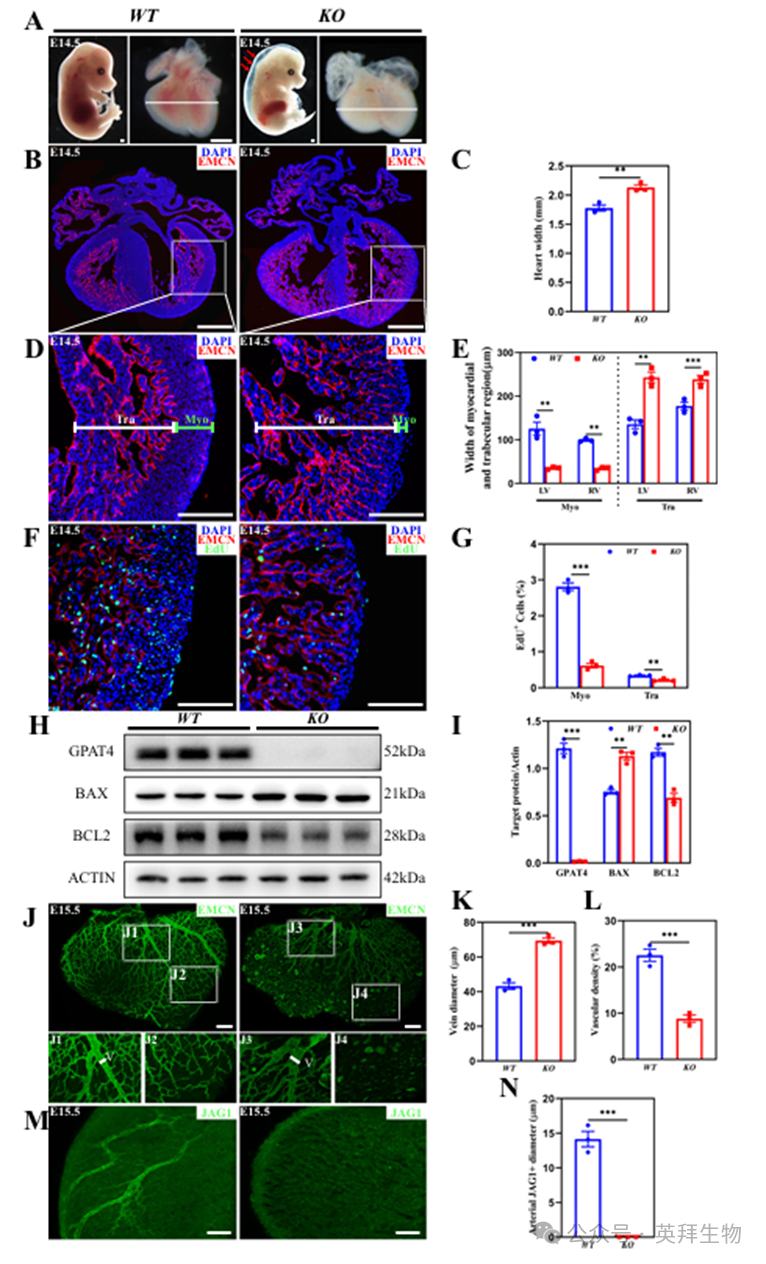
图1.Gpat4基因全球敲除小鼠心脏发育缺陷
2.GPAT4在心内膜发育中的关键作用
接下来,我们构建了Gpat4条件性敲除小鼠(Gpat4 flox)以实现组织特异性基因删除。Gpat4全身敲除小鼠中出现的过度小梁化和心肌层变薄现象提示存在心内膜缺陷。鉴于心内膜细胞属于特殊内皮细胞,我们采用内皮细胞特异性Cre小鼠(Tie2-Cre)对Gpat4基因进行删除。与全身敲除小鼠类似,内皮细胞特异性敲除Gpat4的小鼠(Tie2-Cre; Gpat4F/F)无法存活,并在胚胎发育第14.5天(E14.5)出现显著皮下水肿和心脏扩大(图2A、S2A、B)。组织学分析和免疫荧光染色显示心脏扩张,并证实成功从敲除小鼠心内膜细胞中移除了Gpat4(图2B-E)。与对照组相比,敲除小鼠心脏的肌小梁层明显增厚,而心肌层变薄(图2D、E)。EdU染色显示Gpat4缺失小鼠心脏细胞增殖减少(图2F、G)。Western blot分析证实Gpat4有效缺失,并提示敲除小鼠心脏细胞凋亡增加(图2H,I)。此外,敲除小鼠心脏血管结构呈现异常(图2J-N)。上述结果与全身敲除小鼠的观察结果一致。
此外,我们利用Nfatc1-Cre小鼠特异性敲除心内膜细胞中的Gpat4基因,观察到与全身性及内皮细胞特异性敲除小鼠相似的表型(图2O–R)。
最后,通过Tie2-Cre; Gpat4F/F小鼠心室组织进行离体心脏培养实验发现,与对照组相比,心内膜细胞的外向迁移能力显著降低(图S2C–G)。HUVECs刮痕实验证实敲低Gpat4会损害细胞迁移能力(图S2H,I)。
综合上述结果,证实了GPAT4在心内膜发育中的关键作用。
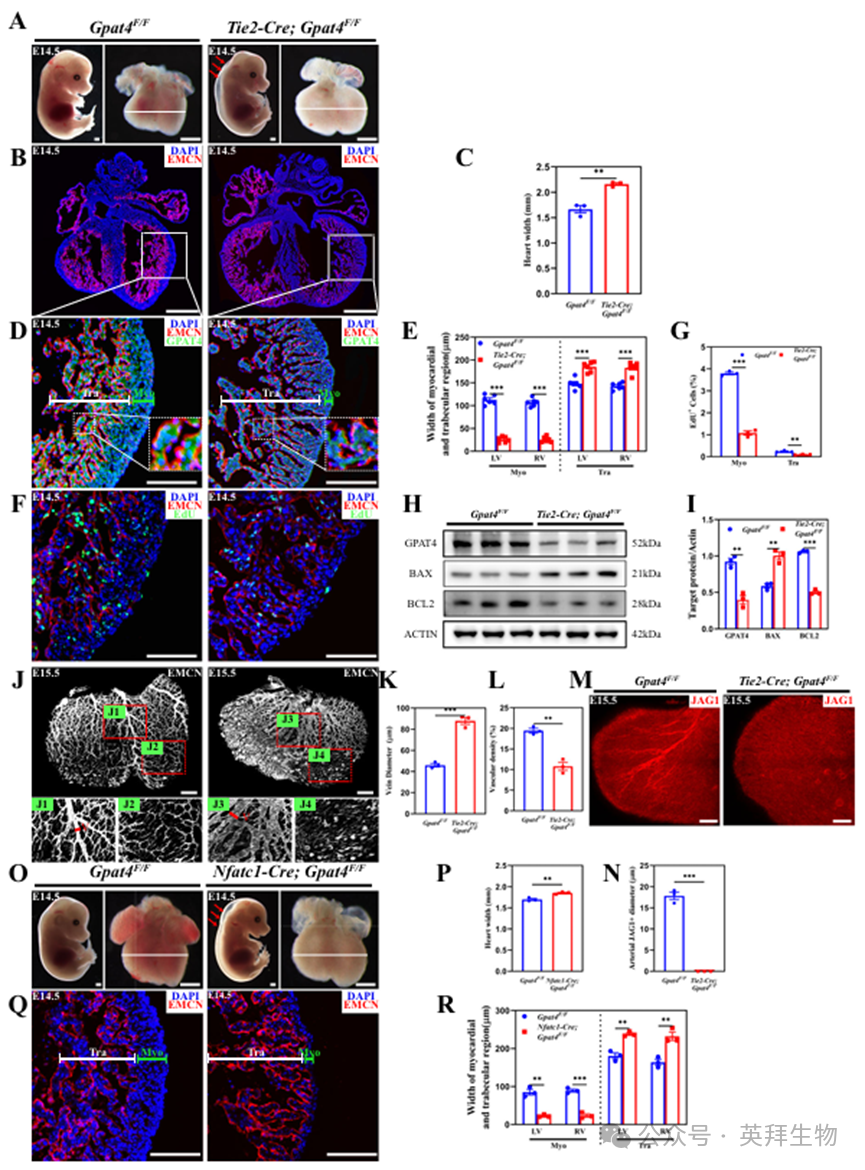
图2.GPAT4在心内膜发育中的关键作用
3.GPAT4缺陷诱发内质网应激反应并增强内质网-线粒体间通信
GPAT4是脂肪细胞和肝细胞中甘油三酯合成的限速酶,GPAT家族包含四个成员:GPAT1-4。在心脏和整个胚胎中检测了甘油三酯(TAG)水平以及其他磷脂(包括磷脂酰胆碱(PC)、PE和磷脂酰肌醇(PI)),但对照组与Gpat4敲除小鼠之间未检测到显著差异(图S3A–C)。心脏组织RNA测序和qPCR分析表明,Gpat4缺失后Gpat1、Gpat2和Gpat3的mRNA水平未发生改变(图S3D-G)。这些发现排除了Gpat4通过调节磷脂合成影响心脏发育的可能性。
对E14.5胚胎心脏组织进行的RNA测序分析揭示,参与内质网应激反应的基因表达显著上调,包括Eif2ak3(编码蛋白激酶RNA样内质网激酶PERK)、热休克蛋白90β家族成员1(Hsp90b1)、Atf4、Atf6及Xbp1(图3A)。这些变化通过qPCR和Western blotting分析得到验证(图3B-D)。
上述结果表明GPAT4在维持内质网稳态中具有关键功能。为深入理解这一机制,我们采用GPAT4抗体进行免疫沉淀(IP),并通过质谱分析在人脐静脉内皮细胞系(HUVEC)中筛选GPAT4结合蛋白,最终鉴定出约30种潜在的GPAT4结合蛋白。随后我们对这些蛋白质参与的生物过程(BP)、所在的细胞区室(CC)以及所执行的分子功能(MF)进行了生物信息学分析。结果揭示这些蛋白质参与蛋白质折叠与重折叠过程,主要定位于内质网,其功能包括结合错误折叠或未折叠蛋白质,并在内质网中处理蛋白质(图3E、F)。其中部分蛋白质属于热休克蛋白70家族(HSPA1A、HSPA1B、HSPA1L和HSPA6),另一些则是内质网应激反应的重要调控因子(EIF2S1/eIF2α、PDIA4和P4HB)(图3F)。
随后我们探究了GPAT4调控UPR的机制。通过采用AlphaFold 3(AF3)——一种能预测几乎所有分子类型复合物形成的精准建模工具——我们识别出EIF2S1/eIF2α与GPAT4之间存在多个结合位点(图3G)。后续免疫沉淀及Western blot实验证实了这两种蛋白质的相互结合(图3H)。
目前普遍认为,PERK对EIF2S1/eIF2α的磷酸化调控着内质网应激引发的未折叠蛋白反应(UPR)。通过整合PERK、GPAT4和EIF2S1/eIF2α三种蛋白的数据,AF3模型揭示了PERK与EIF2S1/eIF2α、GPAT4与EIF2S1/eIF2α同时存在相互关联。然而,AF3并未预测到PERK与GPAT4存在直接关联(图3I)。当从三蛋白组装中剔除GPAT4后,AF3预测PERK与EIF2S1/eIF2α的结合能力显著增强(图3J),相较于GPAT4存在时的结合能力(图3I)。类似地,AF3预测GPAT4与EIF2S1/eIF2α的结合强度在PERK缺失时显著增强(图3G)。这些结果表明GPAT4与PERK在结合EIF2S1/eIF2α时存在竞争关系。
免疫沉淀-Western blot分析显示,敲低GPAT4后PERK与EIF2S1/eIF2α的结合强度显著高于对照组(图3K)。与之相符的是,GPAT4过表达(OE)显著削弱了PERK与EIF2S1的结合(图3L)。结果显示,GPAT4过表达后UPR通路的激活程度大幅降低,磷酸化EIF2S1/eIF2α、CHOP和ATF4的表达水平显著下降(图3M、N)。
通过透射电子显微镜(TEM)对心内膜细胞的进一步观察显示,Tie2-Cre; Gpat4F/F小鼠存在明显的内质网肿胀和线粒体异常(图3O)。此外,在Tie2-Cre; Gpat4F/F小鼠的心内膜细胞中观察到显著增强的内质网-线粒体(ER-Mito)接触(MAMs)(图3O、P)。内质网-线粒体接触通过钙转运通道IP3R-GRP75-VDAC1-MCU和MFN2连接。Western blotting分析显示,与对照组相比,Gpat4突变小鼠心脏组织中这些蛋白质的水平显著升高(图3Q、R)。
综上所述,这些结果表明GPAT4对维持内质网稳态至关重要,其功能受损会引发内质网应激反应,导致未折叠蛋白反应(UPR-ER)增强及内质网-线粒体通信增强。
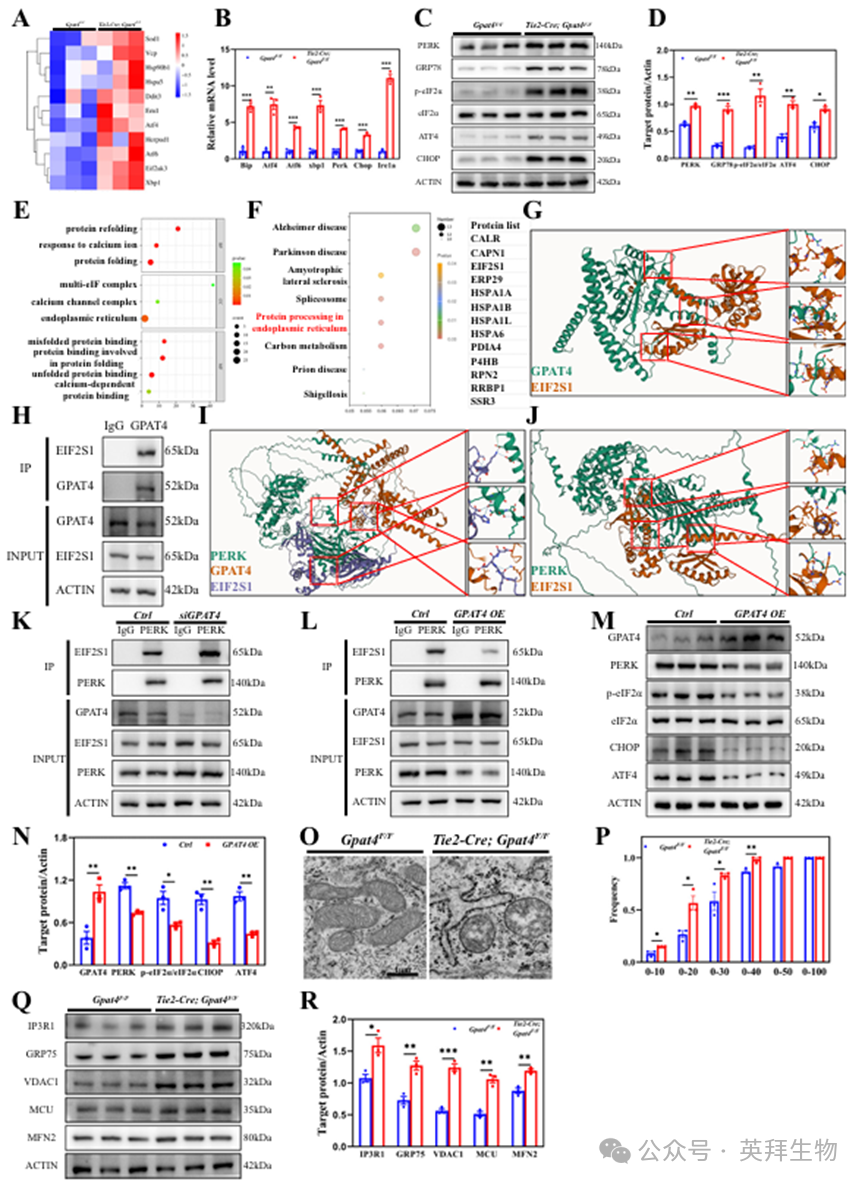
图3.GPAT4调节心内膜中内质网稳态及内质网-线粒体间通信
4.Gpat4缺失诱发cGAS-STING和I型干扰素反应
基于E14.5小鼠心脏组织进行的RNA测序数据集KEGG和GO分析表明,在Tie2-Cre; Gpat4F/F小鼠中与I型干扰素反应相关的通路显著富集(图4A、B)。在表达上调的基因中,许多是干扰素刺激基因(ISGs),包括Usp18、Isg15、Ifit1、Ifna4、Stat1和DDX5825。这种改变的基因表达谱模式在Gpat4全球敲除小鼠的心脏组织(E12.5和E14.5阶段)中也得到了定义(图S4A-F)。qPCR分析证实了这些发现(图4C、S4G、H)。
I型干扰素反应的一个重要触发因素是cGAS-STING-TBK1-IRF3信号通路的激活。我们通过蛋白质印迹法检测该信号通路,结果表明在内皮细胞特异性或全身性Gpat4敲除小鼠的心脏组织中,cGAS、STING、磷酸化TBK1和磷酸化IRF3的蛋白水平均显著升高(图4D、E、S4I、J)。免疫荧光染色显示,增强的cGAS和STING蛋白定位于心内膜(图4F-I)。
我们注意到内质网应答的激活早于cGAS-STING通路的激活(图S4K–N)。在E11.5阶段,Gpat4突变小鼠心内膜中ATF4的表达显著增强(图S4K,L),而cGAS在这些小鼠中几乎无法检测到(图S4M,N)。此外,E14.5心脏组织中显著上调基因数量远多于E12.5心脏(图S4B、C、E、F),该差异通过表达倍数变化得到验证(图S4G、H)。E14.5心脏组织的表达倍数变化同样远高于E12.5心脏组织。
STING作为GPAT433定位于内质网膜。GPAT4的IP-MS结果未检测到Sting蛋白。免疫沉淀实验未发现GPAT4与STING存在直接相互作用(图S5A)。
此外,我们检测了心肌细胞特异性Gpat4敲除小鼠心脏组织中的cGAS-STING通路,未发现对照组与敲除组存在差异(图S5B-D)。
综合上述发现表明,Gpat4功能缺失触发了cGAS-STING信号通路,导致心内膜中I型干扰素反应激活。这些事件发生于内质网应激反应激活之后。
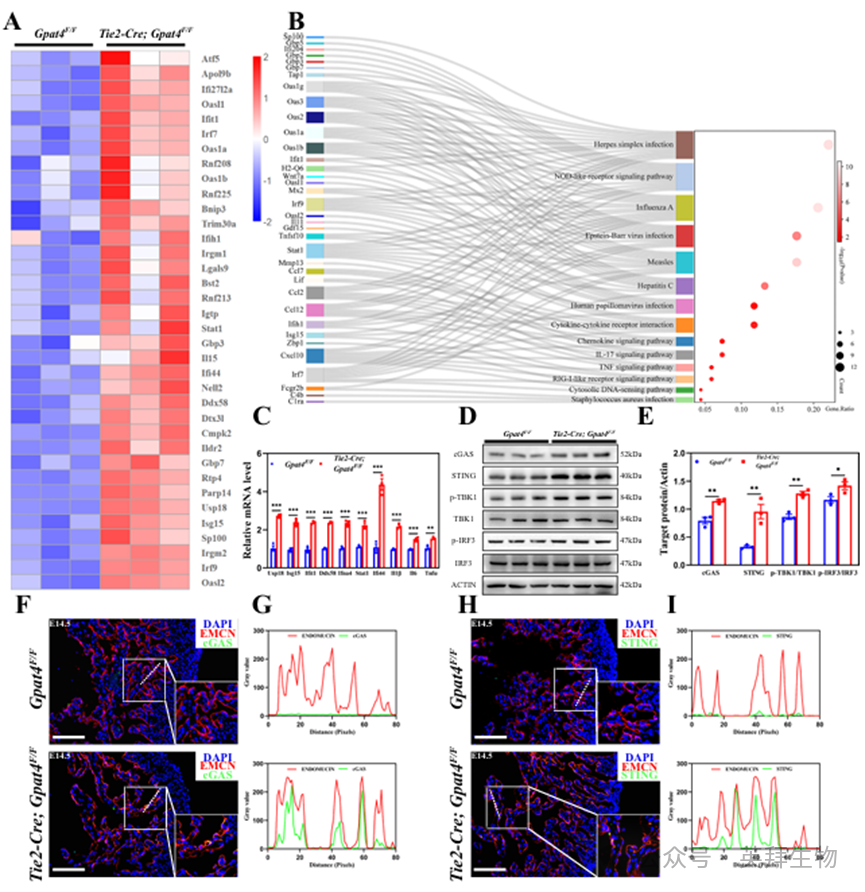
图4.Gpat4缺失诱发心内膜中cGAS-STING-TBK1-IRF3-I型干扰素反应
5.内皮细胞中GPAT4的缺失导致内质网-线粒体通讯增强、线粒体功能障碍以及cGAS-STING通路的激活
接下来,我们在人脐静脉内皮细胞(HUVEC)中开展实验,探究Gpat4敲低与cGAS-STING通路激活之间的关联。通过小干扰RNA(siRNA)技术实现Gpat4敲低。结果显示Gpat4缺失显著损害细胞存活能力(图5A)。Western blot分析显示,敲低Gpat4后促凋亡蛋白BAX、细胞色素C(Cyto-c)及裂解型Caspase-3水平显著升高,而抗凋亡蛋白BCL2水平明显降低(图5B、C)。qPCR和Western blot分析表明SiGpat4细胞中内质网应激反应增强(图5D-F)。内质网-线粒体接触复合体组分(IP3R、GRP75、VDAC1、MCU和MFN2)的蛋白水平在Gpat4敲低细胞中显著升高(图5G、H)。免疫荧光染色显示内质网与线粒体间距离显著缩短(图5I),同时通过荧光MERCs(线粒体-内质网接触位点)报告基因检测(内质网接近线粒体时产生功能性GFP)发现线粒体-内质网接触位点(MAMs)数量增加(图5J)。Gpat4功能缺失还导致cGAS-STING信号通路过度激活及I型干扰素反应增强(图5K-M)。
荧光钙探针(Rhod-2,监测线粒体钙离子)和OMM-NEMOs-mKate-ER(一种新开发的用于测量内质网-线粒体接触区钙离子的指示剂)分析显示,SiGpat4细胞中线粒体及内质网-线粒体接触区的钙信号明显强于对照组(图5N、O)。敲低Gpat4后,线粒体膜电位(使用TMRE和JC-1探针测定)降低,而MitoSox信号(线粒体产生的超氧化物)显著增强(图5P-S)。双链DNA与TOM20的共染色显示,敲低Gpat4后细胞质中泄漏的mtDNA显著增多(图5T)。此外,细胞质mtDNA含量测定表明GPAT4敲低细胞中该含量显著升高(图5V)。线粒体外膜孔道蛋白(VDAC)作为线粒体膜蛋白构成mtDNA释放通道。VDAC寡聚化抑制剂VBIT-4可抑制mtDNA释放。我们观察到VBIT-4处理后干扰素刺激基因(ISGs)表达水平较未处理细胞显著降低(图5W),进一步支持mtDNA释放激活cGAS-STING信号通路的结论。
在心肌细胞系H9c2中,降低GPAT4水平对促凋亡和抗凋亡蛋白影响甚微(图S6A、B)。对照组与SiGpat4细胞间的内质网应激反应及cGAS-STING通路均无显著差异(图S6C、D)。
这些体外研究共同表明,GPAT4的缺失导致内皮细胞中特异性增强的内质网-线粒体通讯、线粒体功能障碍以及cGAS-STING通路的激活。
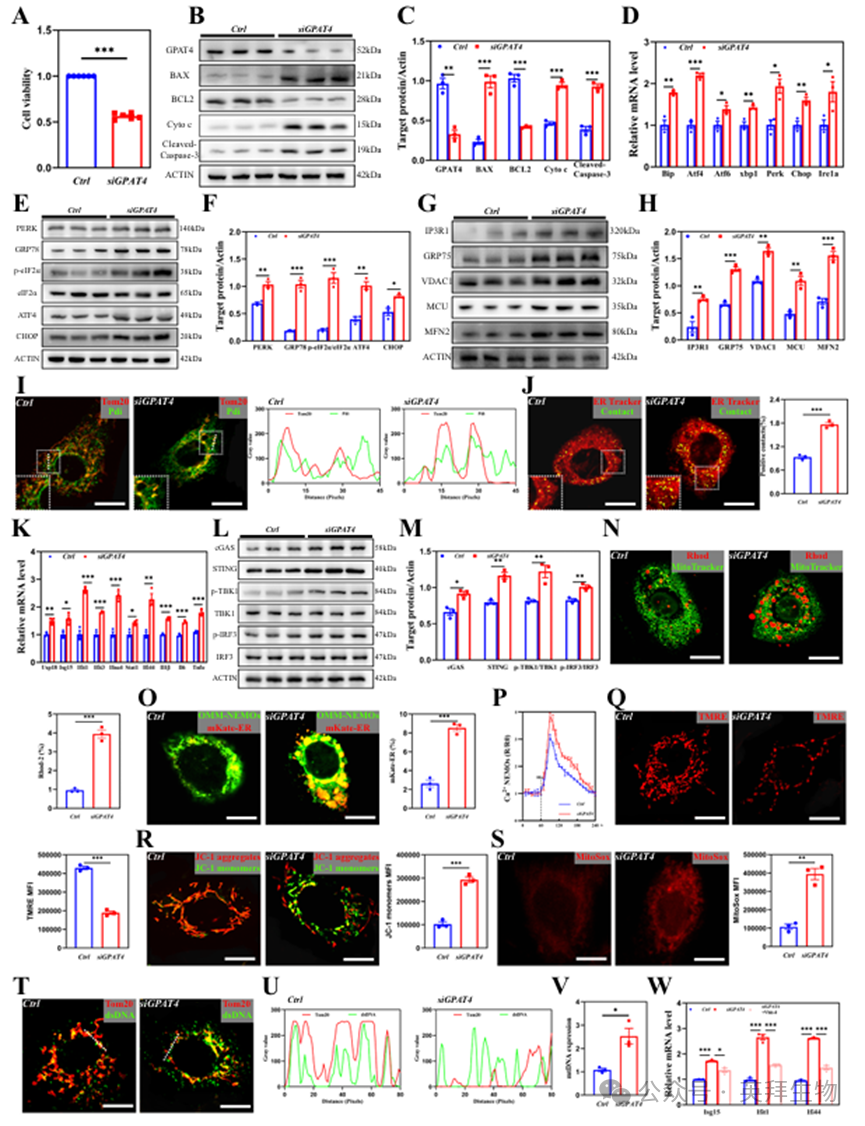
图5.内皮细胞中Gpat4的缺失导致细胞存活能力受损、内质网应激激活、增强内质网-线粒体间通讯并激活cGAS-STING通路
6.阻断cGAS-STING通路挽救了Gpat4缺陷小鼠的心内膜缺陷
为验证阻断cGAS-STING通路能否改善Gpat4敲除小鼠心内膜发育,我们构建了Gpat4/Ifnar1双重敲除小鼠。Ifnar1编码干扰素α和β受体1(IFNAR1),该受体介导I型干扰素反应。在胚胎发育第14.5天(E14.5),Gpat4-/-; Ifnar1-/-(双敲除)小鼠未出现皮下水肿或心脏扩张(图6A、B)。此外,双敲除小鼠的心内膜和心肌发育与对照组相当(图6C、D)。其内质网应激反应与Gpat4敲除小鼠相似(图6E)。然而,与Gpat4-/-小鼠相比,DKO小鼠中I型干扰素信号通路下游基因表达显著下降(图6F)。蛋白水平检测显示,E14.5期DKO小鼠心脏中IFNAR1表达明显降低,且凋亡相关蛋白BAX与BCL2的表达变化趋势与Gpat4-/-小鼠相反(图6G,H)。此外,我们还培育了Gpat4/Sting双敲除小鼠(Gpat4-/-; Sting-/-),该品系未出现水肿或心室扩张,心脏发育与对照组相似(图6I-L)。在mRNA水平上,DKO小鼠的内质网应激反应与Gpat4 KO小鼠相似,而I型干扰素信号通路下游基因的表达水平显著降低(图6M、N)。在蛋白质水平上,与Gpat4-/-小鼠相比,凋亡相关蛋白呈现相反的变化(图6O、P)。
这些发现共同表明,阻断cGAS-STING通路对Gpat4敲除小鼠的心脏发育具有益处,并进一步证实心内膜缺陷是由I型干扰素反应过度激活所导致的。
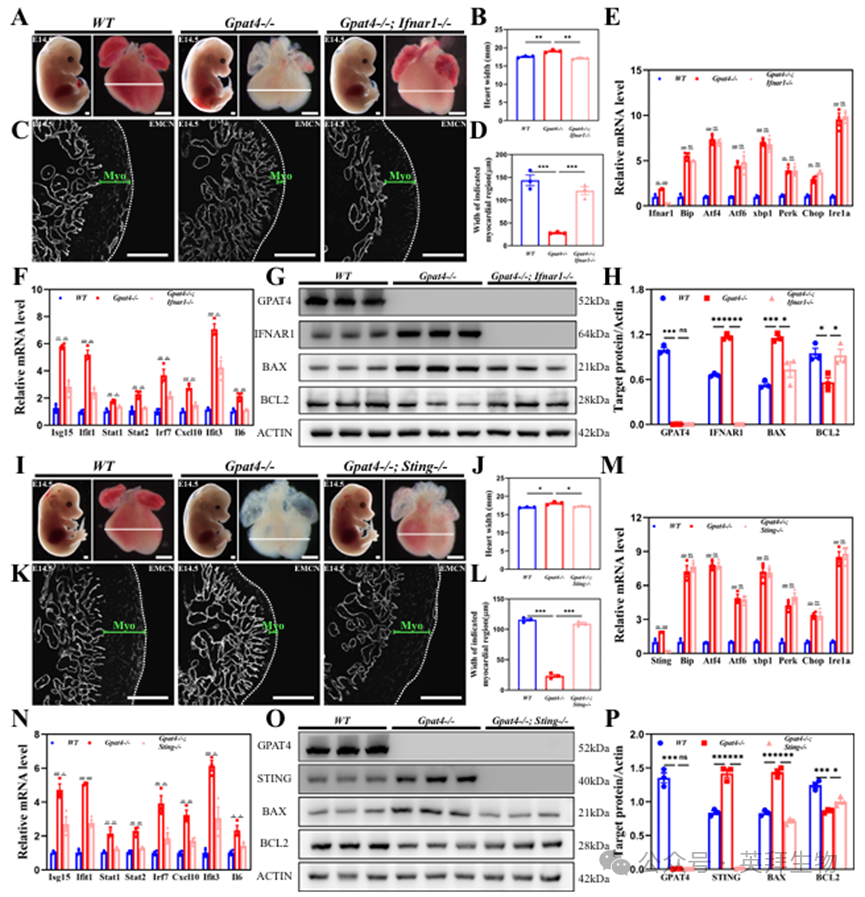
图6.阻断cGAS-STING-I型干扰素反应通路恢复了Gpat4基因缺失小鼠的心内膜发育
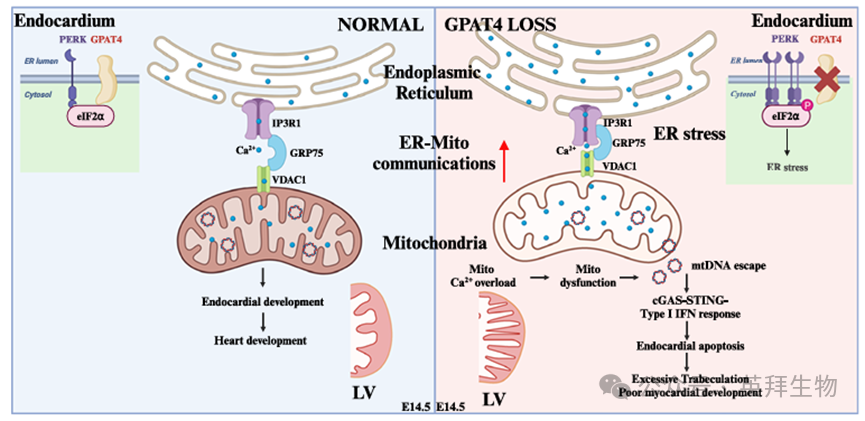
图7.工作模型
结论
综上所述,本研究阐明了Gpat4在胚胎心脏发育过程中对心内膜细胞的复杂调控作用。这些发现有助于深入理解心脏发育的分子机制,并为针对心脏发育异常的潜在治疗策略奠定了基础。
参考文献
Zhao T, Jin K, Wang X, Su X, Wang Y, Gao M, Luo W, Yang H, Yang Z. GPAT4 sustains endoplasmic reticulum homeostasis in endocardial cells and safeguards heart development. Nat Commun. 2025 Apr 8;16(1):3345. doi: 10.1038/s41467-025-58722-5. PMID: 40199910; PMCID: PMC11978851.