






靶向肿瘤–基质间CCL5/CCR5轴以克服神经内分泌前列腺癌的顺铂耐药性
神经内分泌前列腺癌(NEPC)具有高度恶性和快速进展特征,常对传统治疗耐药。尽管顺铂是其一线化疗药,但耐药问题严重限制疗效。研究团队通过系统体内外实验首次发现,CCL5/CCR5 轴是介导 NEPC 顺铂耐药的关键肿瘤–基质对话通路。顺铂诱导的 DNA 损伤可激活癌相关成纤维细胞(CAFs)中的 cGAS-STING 通路,促使其衰老并分泌 CCL5;该因子与肿瘤细胞表面 CCR5 结合后,形成 CCR5/β-arrestin1/p85 复合物,激活 PI3K/AKT 信号通路,从而增强 DNA 修复能力并促使肿瘤逃避凋亡。更重要的是,团队证实 FDA 已批准的 CCR5 拮抗剂马拉维若(maraviroc)可有效阻断此通路,显著提升顺铂对 NEPC 的杀伤效应。在细胞系、患者来源类器官及小鼠模型中,联合用药显著延缓肿瘤生长、延长生存期,且未增加毒性。该研究不仅揭示了 NEPC 顺铂耐药的新机制,还提出了马拉维若“老药新用”的可行策略,为 NEPC 临床化疗优化提供了重要依据。这篇文章于2025年10月发表于《JOURNAL OF EXPERIMENTAL & CLINICAL CANCER RESEARCH》期刊上,IF:12.8。
研究技术路线:

主要实验结果:
1、顺铂可增加癌相关成纤维细胞中CCL5的表达与分泌
为评估神经内分泌前列腺癌(NEPC)的化疗耐药特性,作者利用公认的NEPC小鼠模型(Pb-Cre4: Ptenf/f; Trp53f/f; Rb1f/f)的肿瘤组织建立了同种异体移植模型,该模型可同步肿瘤发生并监测顺铂治疗的反应(图1A)。这些同种异体移植肿瘤始终表现出高度侵袭性和神经内分泌表型,与亲代肿瘤相似。当肿瘤可触及后,将小鼠随机分为顺铂治疗组和PBS对照组,每周治疗一次,共四个周期,模拟临床治疗方案。当PBS组肿瘤达到伦理终点时,收集所有肿瘤并进行RNA测序,以研究顺铂治疗后的变化(图1A)。主成分分析显示,顺铂治疗组与PBS组之间存在显著的转录差异(图1B)。在顺铂组中鉴定出的505个上调基因和339个下调基因中,Ccl5因其折叠变化和P值均表现突出(图1C)。事实上,从顺铂或PBS对照处理后的趋化因子/细胞因子谱分析来看,CCL5表现出高度的组内一致性和显著的组间差异。为验证这一观察结果,作者对同种异体移植肿瘤进行了定量聚合酶链反应(qPCR)和免疫组织化学染色,发现顺铂治疗后Ccl5在RNA和蛋白质水平均上调(图1D和E)。由于难以获得NEPC患者顺铂治疗后的样本,作者改用了先前临床研究中接受新辅助顺铂联合治疗的前列腺腺癌患者的配对样本。治疗前后样本的比较显示,治疗后样本中CCL5的强度和染色百分比均有所增加。综上,这些结果表明顺铂治疗可有效诱导人和小鼠肿瘤样本中CCL5的表达。
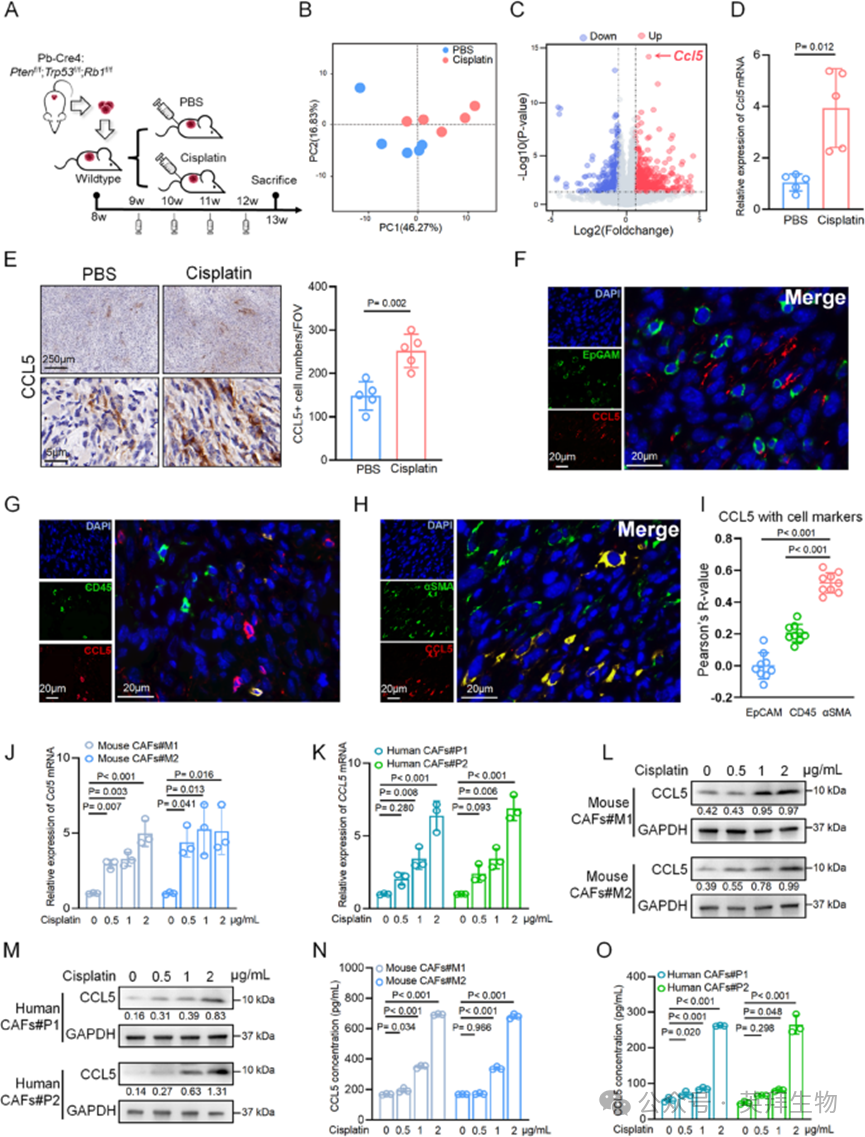
图1 顺铂可增加癌相关成纤维细胞中 CCL5的表达与分泌
确定顺铂治疗可上调CCL5表达后,作者接下来旨在明确负责其上调的特定细胞类型。作者对同种异体移植肿瘤进行了CCL5抗体与多种细胞标志物的免疫荧光共染色。CCL5与α-平滑肌肌动蛋白(αSMA)之间明显且主要的重叠表明,顺铂治疗后癌相关成纤维细胞(CAFs)是CCL5的主要来源(图1F-I)。为进一步证实这一点,作者从NEPC患者和NEPC小鼠的肿瘤组织中分离出CAFs(以下分别称为人CAFs和小鼠CAFs),并在体外用顺铂处理这些细胞。如图1J-M所示,顺铂治疗与CCL5在RNA和蛋白质水平的表达增加趋势相关,且在多个浓度下均观察到上调。酶联免疫吸附试验(ELISA)也表明,顺铂治疗后CAF来源的上清液中CCL5的分泌增加(图1N和O)。综上,这些结果表明顺铂治疗可增加NEPC来源的CAFs中CCL5的表达和释放。
2、顺铂通过cGAS-STING通路诱导CAFs衰老
接下来,作者旨在研究顺铂治疗后CCL5表达增加的上游信号通路。鉴于CCL5是一种关键的衰老相关分泌蛋白,且先前的研究已将顺铂与衰老样表型联系起来,作者推测顺铂诱导CAFs衰老,从而导致CCL5表达增加。对同种异体移植肿瘤RNA测序数据的基因集富集分析(GSEA)显示,顺铂治疗组肿瘤中衰老通路富集(图2A)。为验证这一点,作者对CAFs进行了β-半乳糖苷酶(β-gal)染色,观察到顺铂治疗后β-gal阳性率呈时间和浓度依赖性增加(图2B和C)。相反,从NEPC小鼠中分离的上皮细胞(以下称为小鼠NEPC细胞),即使在高浓度顺铂(2μg/mL)处理下,也未表现出β-gal阳性率增加,反而诱导了细胞死亡。这些结果表明,顺铂优先诱导CAFs衰老,而非肿瘤细胞。
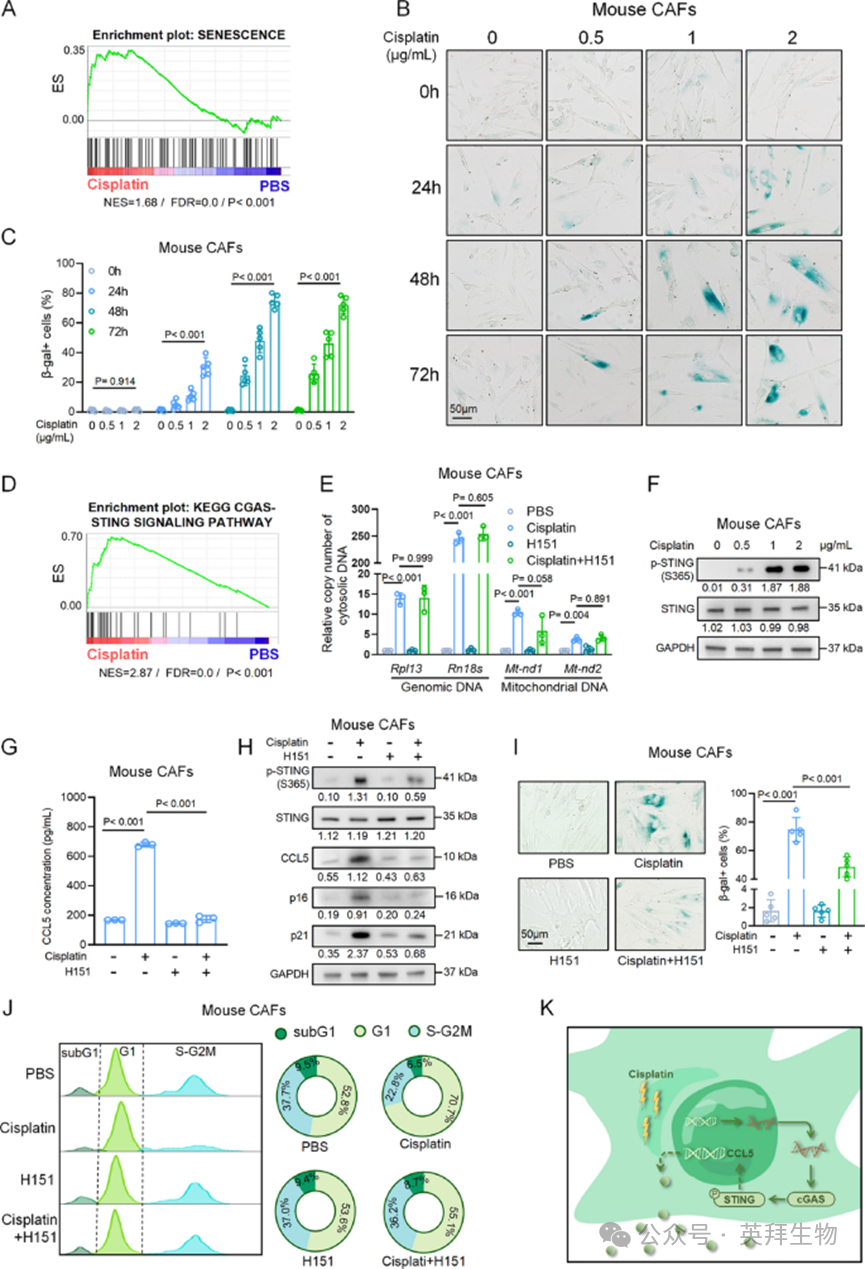
图2 顺铂通过cGAS-STING通路诱导CAFs衰老
作者接下来研究了驱动衰老CAFs中CCL5表达的上游信号通路。基于多项证据,作者推测cGAS-STING通路在调节顺铂诱导的衰老CAFs中CCL5表达中起核心作用:(1)激活的cGAS-STING通路主要通过TANK结合激酶1(TBK1)介导的干扰素调节因子3(IRF3)和核因子κB(NF-κB)激活来调节CCL5转录;(2)cGAS可感知衰老细胞中的胞质DNA片段,这是顺铂诱导的表型;(3)作者的基因集富集分析显示,顺铂治疗的同种异体移植肿瘤中cGAS-STING通路显著富集(图2D)。为验证这一推测,作者评估了CAFs中胞质DNA的释放,结果显示顺铂显著刺激基因组DNA和线粒体DNA释放到胞质中(图2E和S2E)。这种处理还增强了人和小鼠CAFs中STING的磷酸化并增加了CCL5的分泌(图2E-G)。此外,顺铂治疗诱导了CAFs的衰老,表现为p16(Cdkn2a)和p21(Cdkn1a)表达增加、β-gal染色增强以及G1期阻滞(图2H-J)。STING抑制剂H151有效阻断了顺铂诱导的CCL5分泌,并减少了顺铂诱导的衰老表型(图2G-J),尽管它并未减弱胞质DNA的释放(图2E)。综上,这些结果表明,顺铂治疗驱动胞质DNA释放,激活cGAS-STING通路,并诱导CAFs衰老,从而导致CCL5的表达和分泌(图2K)。
3、CAF来源的CCL5介导癌细胞的化疗耐药性
为研究CAF来源的CCL5是否介导肿瘤细胞对顺铂治疗的保护作用,作者首先将肿瘤细胞与来自CAFs的条件培养基共培养,并测试细胞对顺铂治疗的反应(图3A)。从半数抑制浓度(IC50)值和时间依赖性细胞活力来看,顺铂处理的CAFs培养基显著提高了肿瘤细胞的存活率,而未处理的CAFs仅轻微增强了存活率,表明顺铂预处理的CAF来源因子在化疗耐药性中起关键作用(图3B)。
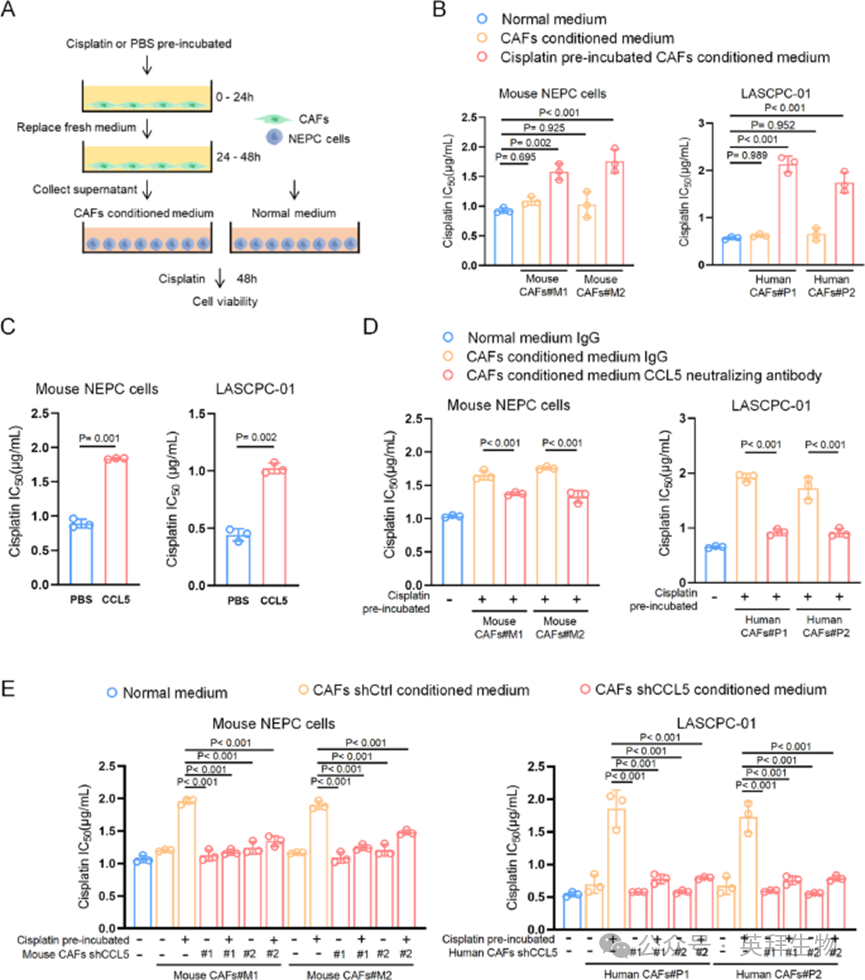
图3 CAFs来源的 CCL5介导癌细胞的化疗耐药性
为确定CCL5是否是造成这种效应的特异性因子,作者补充了重组CCL5,并评估了肿瘤细胞对顺铂的反应。CCL5使人和小鼠肿瘤细胞的IC50值显著增加了约2倍(图3C)。相反,用CCL5中和抗体处理有效消除了CAF来源的条件培养基介导的耐药性(图3D)。作者进一步使用两种独立的短发夹RNA(shRNA)序列在人和小鼠CAFs中敲低CCL5。在顺铂治疗下,CCL5缺陷型CAF来源的条件培养基未增强顺铂耐药性,而野生型CAF来源的培养基则增强了顺铂耐药性(图3E)。这些结果表明,CAF来源的CCL5有效促进了肿瘤细胞对顺铂的耐药性。
4、CCL5/CCR5轴保护癌细胞免受顺铂诱导的DNA损伤和凋亡
为研究CAF来源的CCL5如何调节NEPC细胞对顺铂的耐药性,作者重新分析了同种异体移植肿瘤的转录组数据,发现在顺铂治疗组中与跨膜受体信号相关的通路显著富集(图4A)。CCL5主要通过结合和激活细胞膜受体发挥作用,包括CCR1、CCR3、CCR4和CCR5。为确定负责顺铂耐药性的受体,作者在人和小鼠NEPC细胞中分别敲低了这些受体,并评估了它们对顺铂的反应。在没有CCL5的情况下,所有受体敲低细胞对顺铂的反应与对照细胞相似。然而,添加重组CCL5后,只有CCR5缺陷细胞未能表现出对照细胞中观察到的顺铂耐药性增加(通过IC50测量)(图4B和C)。作者进一步测试了用马拉维若(maraviroc)进行CCR5的药物抑制是否能消除CCL5介导的顺铂耐药性。虽然马拉维若单独使用对有无顺铂存在时的细胞活力影响极小,但它有效抑制了CCL5介导的顺铂耐药性(图4D和E)。这些结果表明,CCR5是介导CCL5保护作用的关键受体。
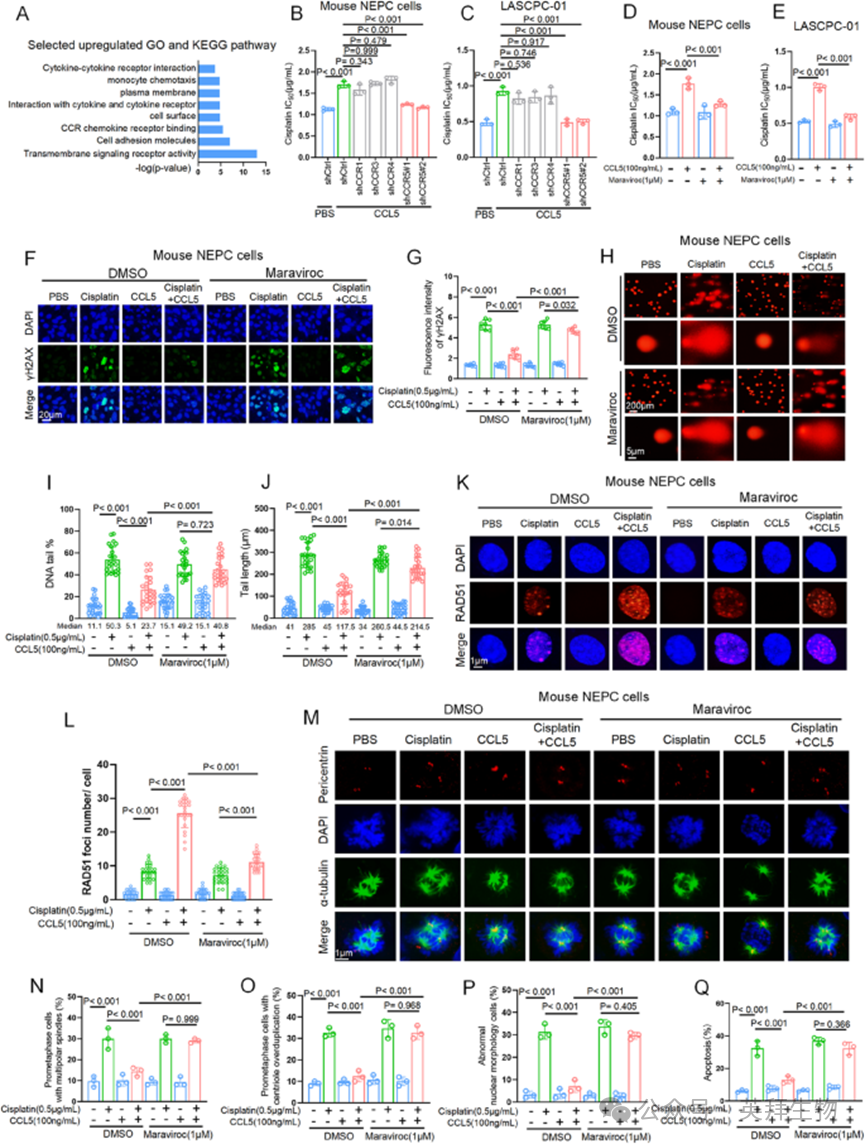
图4 CCL5-CCR5轴保护癌细胞免受顺铂诱导的 DNA 损伤和凋亡
鉴于顺铂主要通过靶向DNA发挥作用,作者通过检测γH2AX灶(DNA双链断裂的敏感报告因子)来测量DNA损伤。如图4F和G所示,重组CCL5处理抑制了顺铂诱导的γH2AX灶形成,表明DNA双链断裂减少。重要的是,这种抑制作用可被马拉维若治疗逆转。碱性彗星试验进一步证实,CCL5保护NEPC细胞免受顺铂诱导的DNA断裂,表现为尾长和尾矩强度降低。同样,马拉维若治疗消除了这种保护作用(图4H-J)。作者接下来通过对小鼠NEPC细胞进行RAD51染色来检查DNA修复能力,结果显示,虽然CCL5在顺铂存在时增加了RAD51灶的形成,但马拉维若有效减弱了CCL5的作用(图4K和L)。作者还检测了增殖细胞核抗原(PCNA)的泛素化,这是另一种DNA修复生物标志物,表明跨损伤合成募集以耐受DNA损伤。然而,尽管顺铂显著诱导了PCNA泛素化,但CCL5未能减弱这种效应,表明CCL5不通过跨损伤合成途径发挥作用。鉴于顺铂诱导的DNA损伤可能引发肿瘤细胞的有丝分裂灾难,作者研究了CCL5是否可保护细胞免受这一过程的影响。作者对小鼠NEPC细胞进行了免疫荧光染色,并检查了微管(α-微管蛋白)、中心体(中心粒周围蛋白)和细胞核(DAPI),以检测微管组织和核形态的任何变化。如图4M-P所示,顺铂治疗显著诱导了有丝分裂灾难,表现为多极纺锤体形成、中心粒过度复制和异常核形态(多核化、微核化、核浓缩和核碎裂)。值得注意的是,CCL5减弱了顺铂对有丝分裂灾难的影响,而马拉维若抵消了CCL5的这种作用,并使肿瘤细胞重新对顺铂敏感。为进一步检查CCL5对顺铂诱导的凋亡的影响,作者用膜联蛋白V/碘化丙啶对小鼠NEPC细胞进行染色,结果表明CCL5有效抑制了顺铂诱导的细胞凋亡(图4Q)。重要的是,顺铂与马拉维若的组合将凋亡恢复到与单独顺铂治疗相当的水平(图4Q)。综上,这些发现强调了CCL5/CCR5通路在保护NEPC细胞免受顺铂诱导的DNA损伤和凋亡中的关键作用。
5、CCL5通过PI3K/AKT通路介导顺铂耐药性
接下来,作者研究了介导NEPC细胞顺铂耐药性的CCL5/CCR5通路的下游信号传导。同种异体移植肿瘤的转录组分析显示,与PBS治疗组相比,顺铂治疗组中PI3K/AKT通路显著上调(图5A)。鉴于AKT在顺铂耐药性中的既定作用及其与CCR信号传导的关联,作者推测CCL5通过CCR5/PI3K/AKT级联反应介导耐药性。免疫荧光染色显示,CCL5处理诱导磷酸化AKT募集到NEPC细胞的质膜(图5B)。在人和小鼠NEPC细胞中,单独的CCL5足以增加AKT的磷酸化,但对细胞外调节蛋白激酶(ERK)、c-Jun氨基末端激酶(JNK)或信号转导和转录激活因子3(STAT3)无影响,突出了PI3K/AKT通路在响应CCL5中的主要作用(图5C和D)。CCL5对AKT的激活抑制了顺铂诱导的γH2AX和裂解的半胱天冬酶3表达,与其对DNA损伤和凋亡的保护作用一致。相反,在CCR5缺陷细胞中,CCL5未能激活AKT或阻止γH2AX和裂解的半胱天冬酶3表达,强调了CCR5在该信号级联反应中的关键作用(图5C和D)。为进一步阐明CCL5触发的转导途径,作者用一系列靶向CCR5(马拉维若)、PI3K(LY294002)和AKT(MK2206)的抑制剂处理人和小鼠NEPC细胞。所有这些抑制剂均有效阻断了CCL5介导的AKT激活(图5E-J)。此外,它们恢复了顺铂诱导的DNA损伤和细胞凋亡,如CCL5处理下γH2AX和裂解的半胱天冬酶3表达所示(图5E-J)。综上,这些发现表明PI3K/AKT通路对于CCL5介导的NEPC细胞顺铂耐药性至关重要。

图5 CCL5通过PI3K/AKT通路介导顺铂耐药性
6、CCL5通过促进CCR5/β-arrestin1/p85复合物的形成诱导AKT磷酸化
作者接下来研究了CCL5/CCR5诱导AKT激活的机制。作为G蛋白偶联受体,CCR5主要通过将细胞外刺激(如CCL5)与特定G蛋白或β-arrestin的激活偶联,从而触发细胞内信号转导。基于先前表征CCR5与β-arrestin或Gi蛋白之间相互作用的结构研究,作者分别敲低了β-arrestin1(Arrb1)、β-arrestin2(Arrb2)以及Gi的关键催化成分,包括Gαi1(Gnai1)、Gαi2(Gnai2)和Gαi3(Gnai3)。仅在β-arrestin1缺失时,CCL5介导的AKT磷酸化被消除(图6A)。这一发现在公认的人NEPC细胞系LASCPC-01中得到证实,其中β-arrestin1消融减弱了CCL5诱导的AKT磷酸化(图6B)。为进一步研究该信号级联反应的分子细节,作者敲低了小鼠NEPC细胞中的每个PI3K亚基,包括p85α(Pik3r1)、p85β(Pik3r2)、p55(Pik3r3)、p110α(Pik3ca)、p110β(Pik3cb)和p110δ(Pik3cd)。敲低p110α或同时敲低p85α和p85β足以抑制CCL5介导的AKT磷酸化(图6C-E)。这些结果强调了PI3K调节亚基p85和催化亚基p110α在转导CCL5信号中的重要性。确定了所涉及的关键PI3K亚基后,作者接下来旨在确定β-arrestin1是否与PI3K相互作用,与CCR5形成信号复合物。先前的研究已报道p85与β-arrestin1之间的相互作用,因此作者推测CCR5、β-arrestin1和p85形成复合物,通过p110α转导CCL5诱导的AKT磷酸化。为验证这一假设,作者在共表达Flag-CCR5、V5-β-arrestin1和HA-p85α/HA-p85β的人胚肾293T(HEK293T)细胞中进行了免疫共沉淀试验。仅在CCL5存在时,CCR5与β-arrestin1和p85α/p85β共免疫沉淀,这也诱导了AKT磷酸化,将复合物与下游信号传导联系起来。在没有β-arrestin1的情况下,这种相互作用被消除(图6F)。随后在人和小鼠NEPC细胞中进行的免疫共沉淀试验证实了CCR5/β-arrestin1/p85复合物的形成(图6G),该复合物可被β-arrestin1敲低破坏(图6H和I)。综上,这些发现表明CCL5促进CCR5/β-arrestin1/p85复合物的形成,这对于响应CCL5的AKT激活至关重要(图6J)。
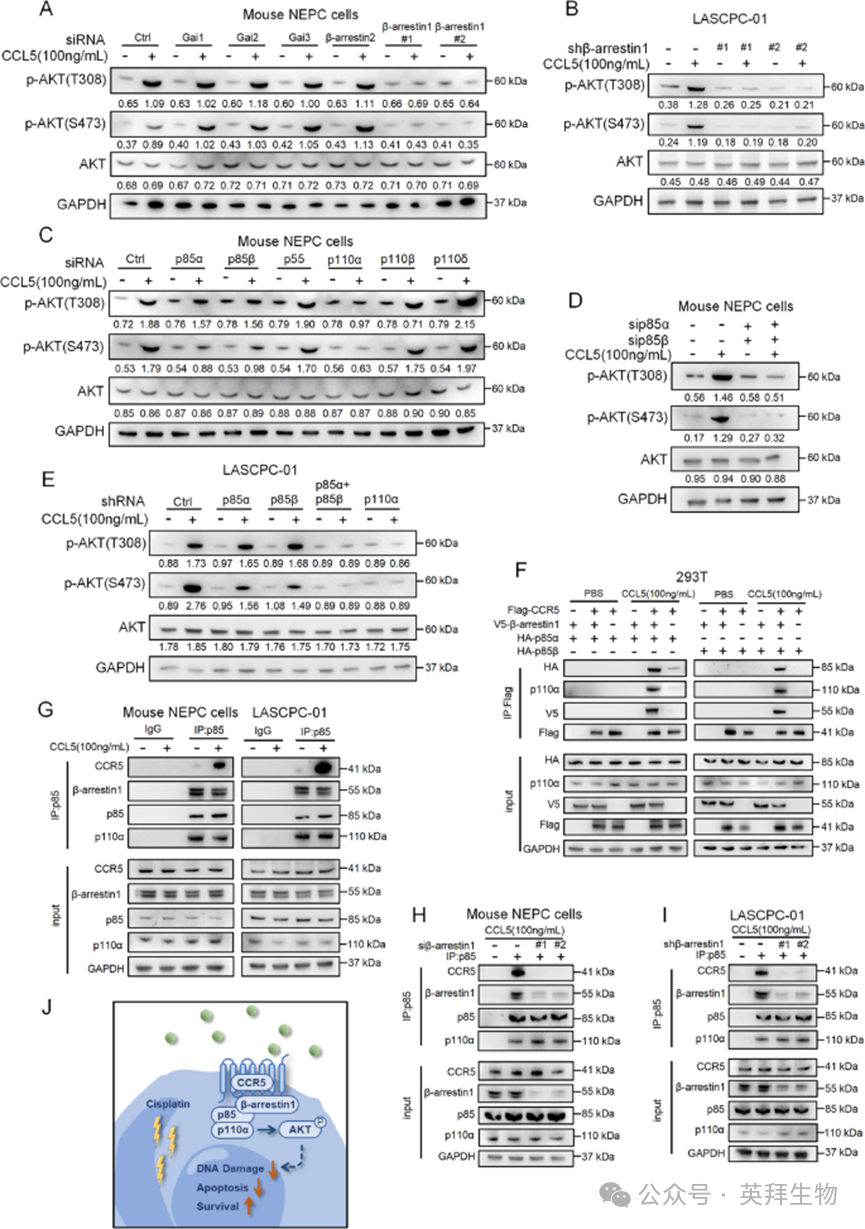
图6 CCL5通过促进CCR5/β-arrestin1/p85的形成诱导AKT磷酸化
7、靶向CCL5/CCR5通路可克服顺铂耐药性并提高化疗效率
上述结果揭示了CCL5/CCR5/β-arrestin1/PI3K/AKT级联反应在顺铂耐药性中的独特作用,并促使作者评估在临床前模型中靶向该通路的治疗潜力。作者首先将小鼠NEPC细胞注射到CCL5敲除小鼠或其野生型同窝小鼠中(图7A)。在没有顺铂的情况下,两组之间的肿瘤生长相当(图7B)。然而,在顺铂治疗下,与野生型对照相比,CCL5敲除受体小鼠的肿瘤生长显著减少(图7B)。组织化学分析显示,在没有顺铂的情况下,敲除组和野生型组之间的肿瘤增殖和凋亡没有差异(通过PCNA和TUNEL染色)(图7C和D)。相反,在顺铂治疗下,来自CCL5敲除小鼠的肿瘤与对照组相比表现出增殖减少和凋亡增加,突出了CCL5在顺铂治疗期间肿瘤微环境中的关键作用(图7C和D)。
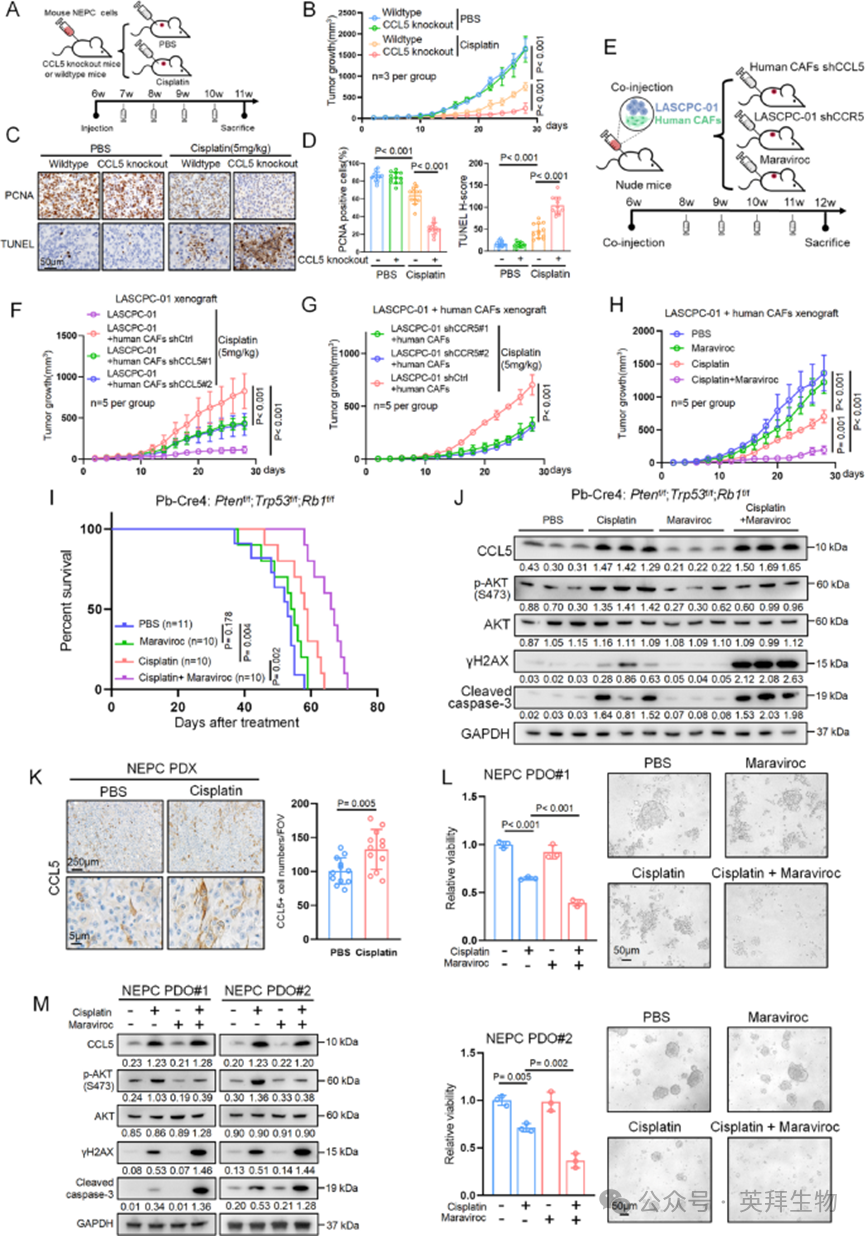
图7 靶向CCL5/CCR5通路可克服顺铂耐药性并提高化疗效率
接下来,作者通过肿瘤-基质相互作用测试了CCL5/CCR5通路对异种移植肿瘤生长的影响。将裸鼠皮下注射LASCPC-01细胞单独或与人类CAFs共注射,随后进行顺铂治疗(图7E)。与单独的LASCPC-01细胞相比,与CAFs共注射显著增强了肿瘤生长(图7F)。当CAFs中的CCL5被敲低时,这种效应被减弱,尽管CCL5消融对异种移植物中的细胞增殖或上皮-成纤维细胞比率没有影响(图7F)。这些发现表明CAF来源的CCL5调节肿瘤细胞对顺铂的反应。支持这一点的是,LASCPC-01细胞中的CCR5敲低显著减少了与CAFs共注射的异种移植物中的肿瘤生长(图7G)。马拉维若在使异种移植物对顺铂治疗敏感方面表现出与CCR5敲低相似的效果,尽管马拉维若单独使用几乎不抑制肿瘤生长(图7H)。神经元特异性烯醇化酶(NSE)、PCNA和TUNEL染色进一步证实了这些结果:虽然马拉维若单独使用效果温和,但其与顺铂的组合抑制了神经内分泌表型、肿瘤增殖并诱导了凋亡。值得注意的是,顺铂、马拉维若及其组合均不影响CCR5表达。
由于马拉维若在小鼠中未表现出明显的毒性作用(如体重稳定所示),作者接下来检查了其在NEPC小鼠中的作用。对8周龄去势小鼠进行PBS、马拉维若、顺铂或两者组合治疗。虽然马拉维若单独使用效果极小,但组合治疗显著延长了总生存期,且不改变体重(图7I)。肿瘤样本的蛋白质印迹分析显示,尽管个体样本之间存在一些异质性,但顺铂适度增加了γH2AX和裂解的半胱天冬酶3(图7J)。顺铂还诱导了CCL5表达和AKT磷酸化,这可能限制其疗效。值得注意的是,马拉维若与顺铂联合使用抑制了AKT磷酸化,从而增强了DNA损伤和凋亡(图7J)。
为进一步验证这些发现,作者从基因修饰的小鼠模型转向了来自NEPC肿瘤的患者来源异种移植物(PDX)。与上述同种异体移植肿瘤相似,顺铂增加了CCL5表达,其在很大程度上与αSMA重叠,表明CAFs是CCL5的主要来源(图7K)。然而,患者来源异种移植肿瘤生长速率的不均匀性限制了对治疗效果的直接评估。相反,作者从患者来源异种移植肿瘤生成类器官,并在体外用PBS、马拉维若、顺铂或其组合处理它们。如图7L所示,顺铂治疗抑制了类器官增殖并减小了类器官大小(通过CellTiter-Glo数据和代表性明场图像反映)。重要的是,与单独使用顺铂相比,顺铂与马拉维若的组合显著增强了抑制效果(图7L)。类器官的蛋白质印迹分析进一步表明,马拉维若抑制了顺铂诱导的AKT磷酸化,且与单独使用顺铂相比,马拉维若和顺铂的组合显著增加了DNA损伤和凋亡(通过γH2AX和裂解的半胱天冬酶3水平升高表明)(图7M)。综上,这些发现表明靶向CCL5/CCR5轴可提高顺铂治疗NEPC的疗效,为进一步的临床开发提供了有力的理论依据。
结论
本研究表明,在NEPC中, CAFs分泌的CCL5可与肿瘤细胞表面的CCR5结合,激活PI3K/AKT通路,从而保护肿瘤细胞免受DNA损伤及顺铂诱导的细胞死亡。CCR5拮抗剂马拉维若(maraviroc)可破坏CCL5/CCR5信号通路,阻断上述保护作用,进而提高顺铂类化疗的疗效。
参考文献
Liu B, Zhang W, Ji Y, Wu J, Su R, Liu X, Li A, Shen K, Chai X, Wu H, Ma Z, Hu C, Jiang Z, Dong L, Zhu Y, Dong B, Xue W, Pan J, Wang Q. Targeting the CCL5/CCR5 axis in tumor-stromal crosstalk to overcome cisplatin resistance in neuroendocrine prostate cancer. J Exp Clin Cancer Res. 2025 Oct 28;44(1):296. doi: 10.1186/s13046-025-03552-y. PMID: 41152972; PMCID: PMC12570517.