






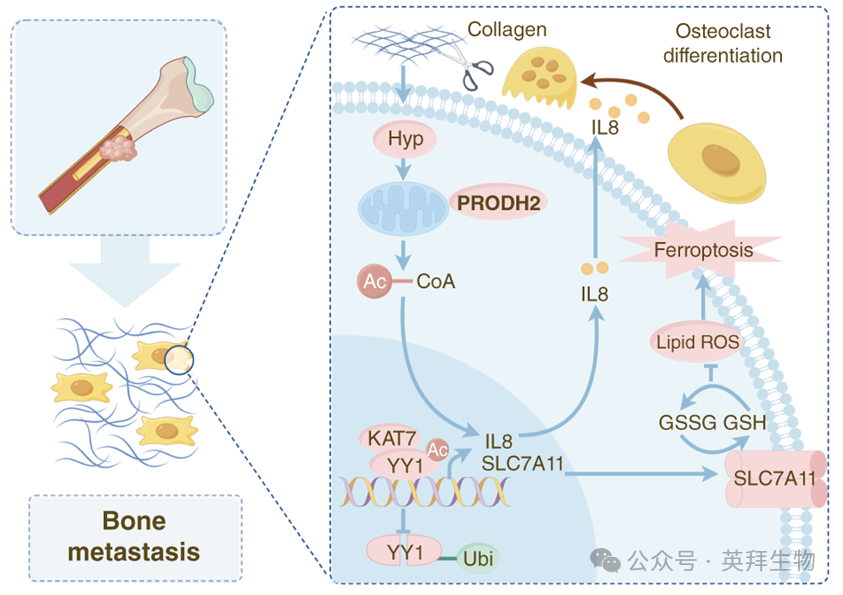
PRODH2介导的骨代谢微环境促进乳腺癌转移
乳腺癌常发生骨转移,但针对骨转移性乳腺癌的治疗手段十分有限。骨转移微环境中的氨基酸代谢被重编程,提示这可能是一个可靶向的薄弱环节。在本研究中,作者聚焦于羟脯氨酸(Hyp)的代谢——这种由骨胶原降解产生的关键氨基酸,是骨转移的重要生物标志物。在临床获取的乳腺癌骨转移样本中,Hyp代谢的核心酶——脯氨酸脱氢酶2(PRODH2)表达显著上调。值得注意的是,PRODH2介导的Hyp代谢可驱动破骨细胞分化,加剧胶原降解,并在体内促进乳腺癌骨转移。此外,PRODH2分别通过上调铁死亡抑制因子SLC7A11和骨转移相关因子CXCL8(IL8),维持肿瘤细胞存活并促进破骨细胞分化。更有趣的是,PRODH2催化Hyp代谢产生的乙酰辅酶A可增强转录因子YY1的乙酰化,从而协同激活SLC7A11和IL8的转录。重要的是,使用PRODH2抑制剂能有效阻断这一骨转移级联反应。综上,骨溶解导致的胶原降解产生Hyp,反过来又强化破骨细胞分化与转移,形成恶性循环。总之,作者的研究揭示的PRODH2–SLC7A11–IL8轴在促进乳腺癌骨转移中的作用,为开发新的治疗策略提供了理论依据。该研究于2025年8月发表在《Cancer Research》,IF 16.6分。
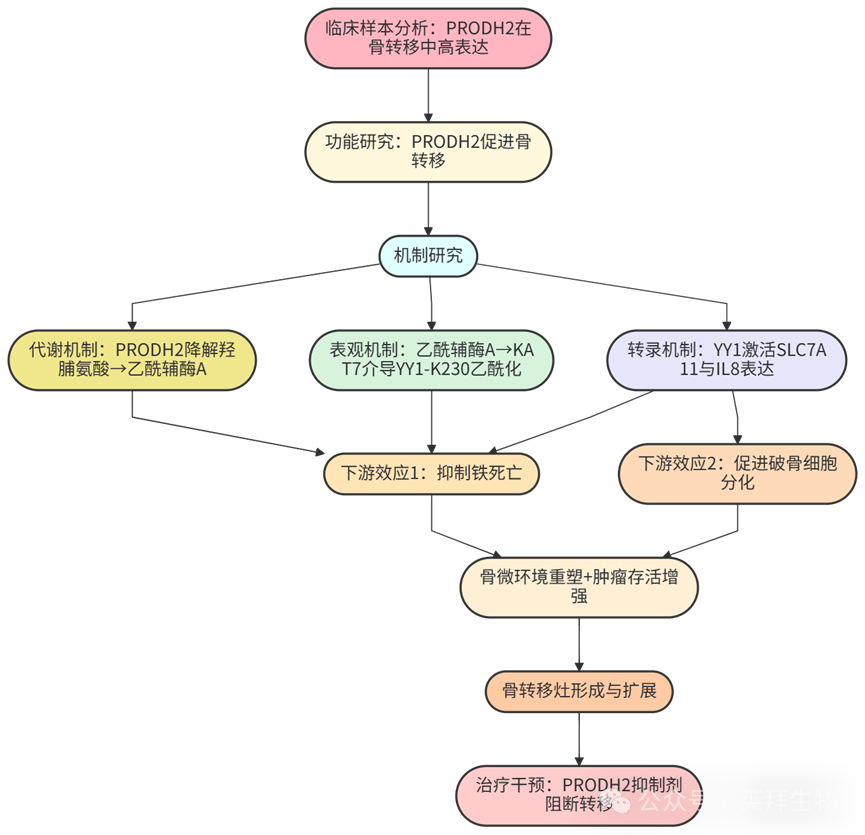
技术路线:
主要研究结果:
1、PRODH2介导的Hyp代谢促进乳腺癌骨转移
鉴于骨胶原在骨基质代谢及转移中的关键作用,以及骨转移会伴随胶原降解并释放羟脯氨酸(Hyp)的事实,作者分析了Hyp代谢关键酶PRODH2在乳腺癌中的分布与功能。免疫组化显示,与原位乳腺癌、非骨转移乳腺癌及正常乳腺组织相比,骨转移灶中PRODH2表达显著升高(图1A)。与之吻合,高骨转移潜能细胞系(JIMT-1、HCC1806、SCP2)的PRODH2水平明显高于低骨转移潜能细胞系(MCF7、SKBR3、MDA-MB-231、4175、4T1)。
为进一步验证功能,作者构建了PRODH2稳定过表达的MDA-MB-231、MCF7细胞,并在高骨转移SCP2细胞中敲除PRODH2(图1B)。体外共培养实验显示,无外源Hyp时PRODH2不影响破骨细胞分化;而加入Hyp后,PRODH2过表达细胞的条件培养基(CM)显著增加多核破骨细胞数量,PRODH2敲除CM则抑制分化(图1C)。
小鼠胫骨注射模型中,PRODH2显著促进乳腺癌细胞在骨微环境中的定植与生长,并伴随更多破骨细胞分化(图1D)。心内注射模型进一步表明,PRODH2敲除显著降低骨转移负荷、减少破骨细胞分化及胶原降解,提高骨小梁相对体积、数量和厚度,缩小骨小梁分离度(图1E–G)。超低吸附实验显示,PRODH2缺失不影响循环肿瘤细胞存活,提示其关键作用发生在骨定植阶段而非血行存活。值得注意的是,PRODH2过表达显著提高胫骨及血清Hyp水平,敲除则相反(图1D、F)。综上,PRODH2通过诱导破骨细胞分化与胶原降解促进乳腺癌骨转移。
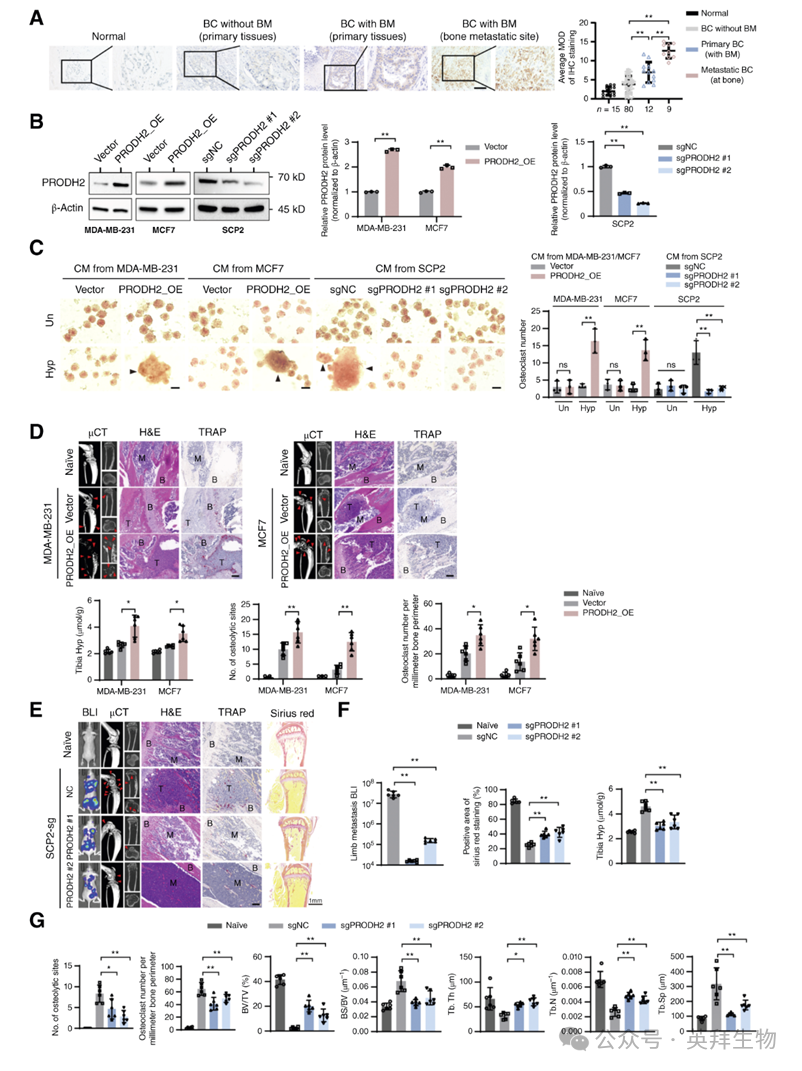
图1:PRODH2介导的低代谢促进乳腺癌骨转移
2、PRODH2通过调控IL8分泌促进破骨细胞分化
为解析机制,作者经4轮体内胫骨筛选建立高骨转移亚系MDA-MB-231-BM4。生物发光成像(BLI)显示,BM4细胞仅需3周即可在骨内形成强信号,而亲本细胞需5周(图2A)。伴随骨趋向性增强,BM4细胞PRODH2表达及胫骨Hyp浓度显著升高。
RNA-seq与GSEA分析发现,BM4细胞中骨重塑因子富集、铁死亡通路受抑(图2B)。qPCR验证显示,IL8在PRODH2过表达及BM4细胞中增幅最大(图2C)。ELISA亦证实,PRODH2过表达CM中IL8分泌升高,敲除则降低(图2D)。临床数据集GSE2603表明,IL8高表达与乳腺癌患者无转移生存期缩短显著相关(r = –0.294,P < 0.01)。
功能实验显示,重组IL8显著增强Hyp诱导的破骨细胞分化,而shRNA或中和抗体敲低IL8则有效逆转PRODH2过表达细胞的促分化效应(图2E)。体内实验中,IL8敲低显著抑制SCP2骨转移、破骨细胞分化及胶原降解,并降低胫骨Hyp水平(图2F、G)。因此,PRODH2通过上调IL8分泌驱动破骨细胞分化,加速乳腺癌骨转移。
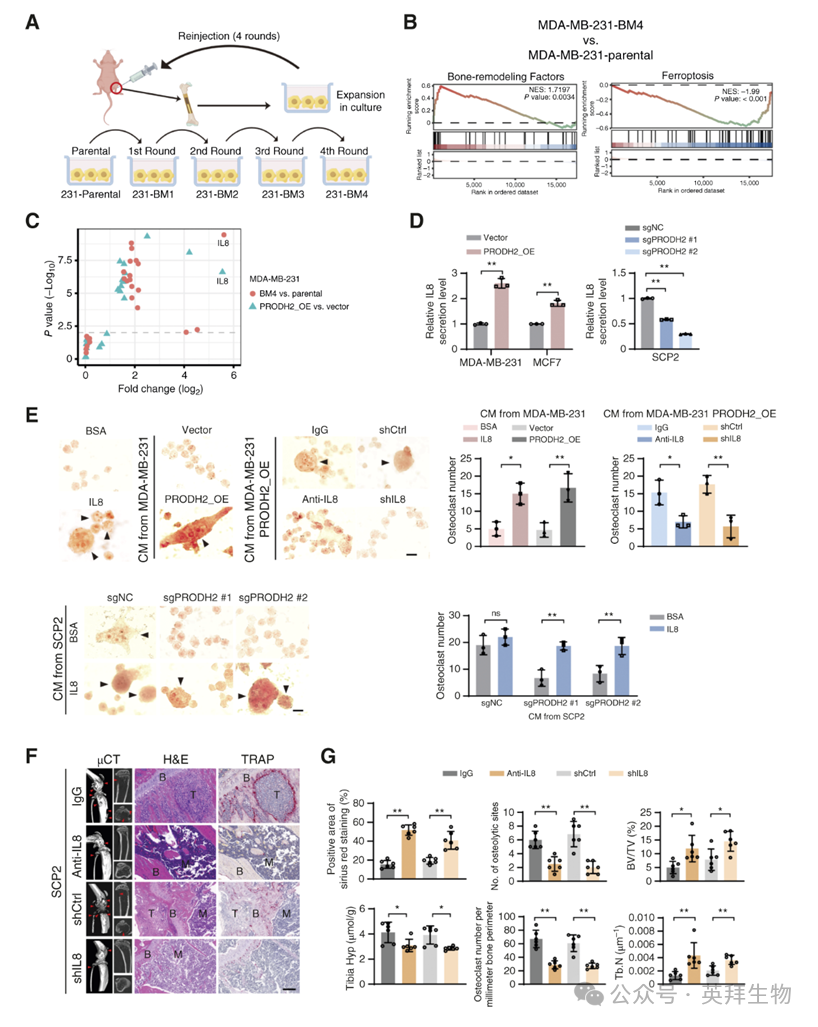
图2:PRODH2调节il - 8分泌,促进破骨细胞分化
3、PRODH2抑制乳腺癌细胞的铁死亡
在骨转移模型中,MDA-MB-231-BM4细胞表现出PRODH2升高并伴随铁死亡受抑,促使作者进一步探究机制。骨转移灶缺氧可通过hepcidin诱导铁过载。作者推测,缺氧且富铁的骨微环境会筛选出天生耐铁死亡的肿瘤亚克隆,其中PRODH2上调正是早期定植、氧化与代谢应激峰值时的关键生存机制。
在骨转移灶中,MDA-MB-231-BM4细胞的经典铁死亡标志物MDA与4HNE水平下降(图3A)。心内注射模型中,仅铁死亡抑制剂Fer-1可逆转PRODH2敲除导致的骨转移抑制,而凋亡、坏死性凋亡及自噬抑制剂均无效。
体外构建肿瘤细胞-破骨细胞共培养体系,加入Ⅰ型胶原后,PRODH2过表达提高Hyp水平并减轻erastin/RSL3诱导的铁死亡;敲除则相反。若无胶原或破骨细胞,Hyp与细胞活性均无显著变化。
进一步证实,Hyp处理仅在PRODH2过表达细胞中增强其对铁死亡诱导剂的抵抗;敲除则失去该效应(图3B)。提示肿瘤细胞-破骨细胞互作促进胶原降解释放Hyp,进而诱导肿瘤细胞耐铁死亡。
增殖曲线显示,无论有无Hyp,PRODH2过表达或敲除均不影响细胞增殖,说明细胞活性与骨定植差异源于铁死亡抵抗而非增殖差异。
机制层面,流式及荧光显微镜示PRODH2过表达显著削弱erastin诱导的脂质过氧化;敲除则加剧(图3C、D)。电镜下,Hyp+erastin处理时,PRODH2过表达抑制线粒体破裂与嵴结构丢失,敲除则加重(图3E)。综上,PRODH2抑制乳腺癌细胞铁死亡。
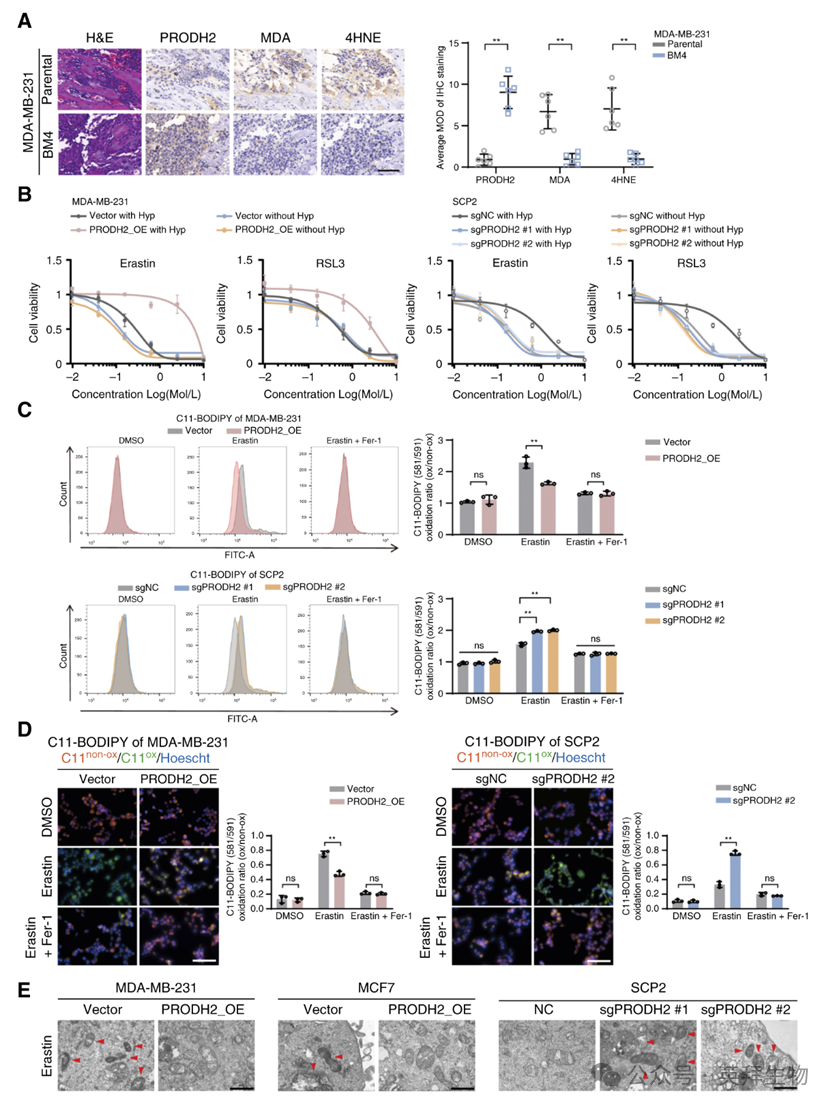
图3:PRODH2抑制乳腺癌细胞铁下垂
4、PRODH2 抑制铁死亡并调控破骨细胞分化
为阐明 PRODH2 介导的铁死亡抑制机制,作者进行了代谢组学分析。在 Hyp 存在下,PRODH2 过表达细胞的 GSH 代谢通路显著富集,谷氨酸与 GSH 水平升高(图 4A)。作为最主要的铁死亡抑制剂,GSH 通过 SLC7A11 依赖的半胱氨酸摄取合成(24)。作者发现,PRODH2 过表达提高胞内 GSH 含量,而敲除则降低其水平(图 4B);且仅调控 SLC7A11 表达,不影响其他铁死亡相关蛋白或 Fe²⁺水平(图 4C、D)。
救援实验显示,在 PRODH2 过表达背景下敲低 SLC7A11 可逆转 GSH 升高(图 4E)、加剧脂质过氧化(图 4F)并抑制破骨细胞分化(图 4G)。相反,于 PRODH2 敲除细胞中过表达 SLC7A11 则恢复上述表型。体内模型进一步证实,sgPRODH2 细胞中回补SLC7A11 可促进破骨细胞生成与胶原降解(图 4H)。因此,PRODH2 通过上调 SLC7A11、增加 GSH 合成来抵抗铁死亡。
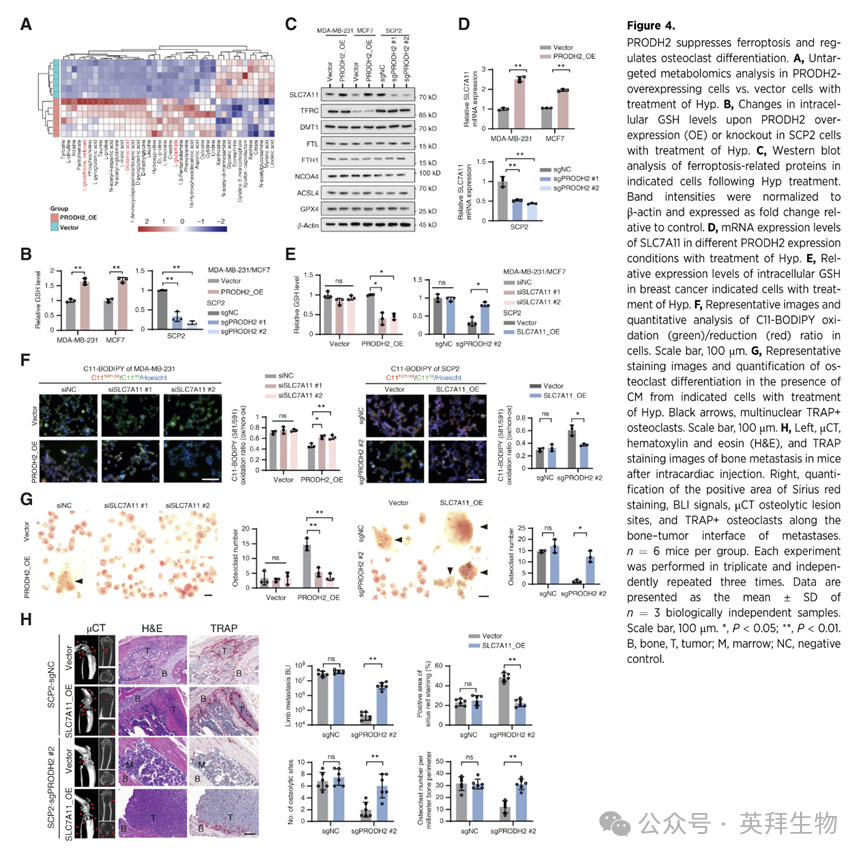
图4:PRODH2抑制铁下垂,调节破骨细胞分化
5、PRODH2-Hyp 通过增强 YY1 乙酰化促进骨转移
肿瘤细胞-破骨细胞共培养(+Ⅰ型胶原)体系显示,PRODH2 过表达协同上调 SLC7A11 与 IL8 的转录及蛋白水平,敲除则相反。为解析其机制,作者采用 CAPTURE-质谱筛选 SLC7A11/IL8 启动子结合蛋白,并经 qRT-PCR 与ChIP 验证 YY1 及其乙酰转移酶 KAT7 可直接结合两基因启动子并调控转录(图 5A–C)。
已知 YY1 的转录活性受其磷酸化、乙酰化、泛素化等修饰调节。作者发现,PRODH2 过表达显著提高胞内乙酰辅酶 A 与丙酮酸水平,而催化失活突变体 Y446A 无此效应,提示酶活性为乙酰辅酶 A 生成所必需。乙酰辅酶 A 介导赖氨酸乙酰化,动态调节蛋白功能。共免疫沉淀-质谱及原位 PLA 显示,PRODH2 增强 KAT7 与 YY1 的结合(图 5E–G),并促进 YY1-K230 位乙酰化(图 5H)。失活突变体丧失该能力,而外源乙酰辅酶 A 可恢复 YY1-K230 乙酰化及其转录活性。
此外,PRODH2 延长 YY1 蛋白半衰期,而 K230R 突变缩短之(图 5I)。机制上,PRODH2 通过 KAT7 介导的乙酰化抑制 YY1 泛素化降解(图 5J);K230R 或 siKAT7 均增加 YY1 泛素化(图 5K)。双荧光素酶报告实验表明,K230R 抑制 PRODH2 介导的 YY1 转录活性,而 K230Q(乙酰化模拟)维持其持续激活(图 5L)。综上,PRODH2 以乙酰辅酶 A 依赖方式促进 KAT7 对 YY1-K230 的乙酰化,抑制其泛素化降解,从而协同上调 SLC7A11 与 IL8。
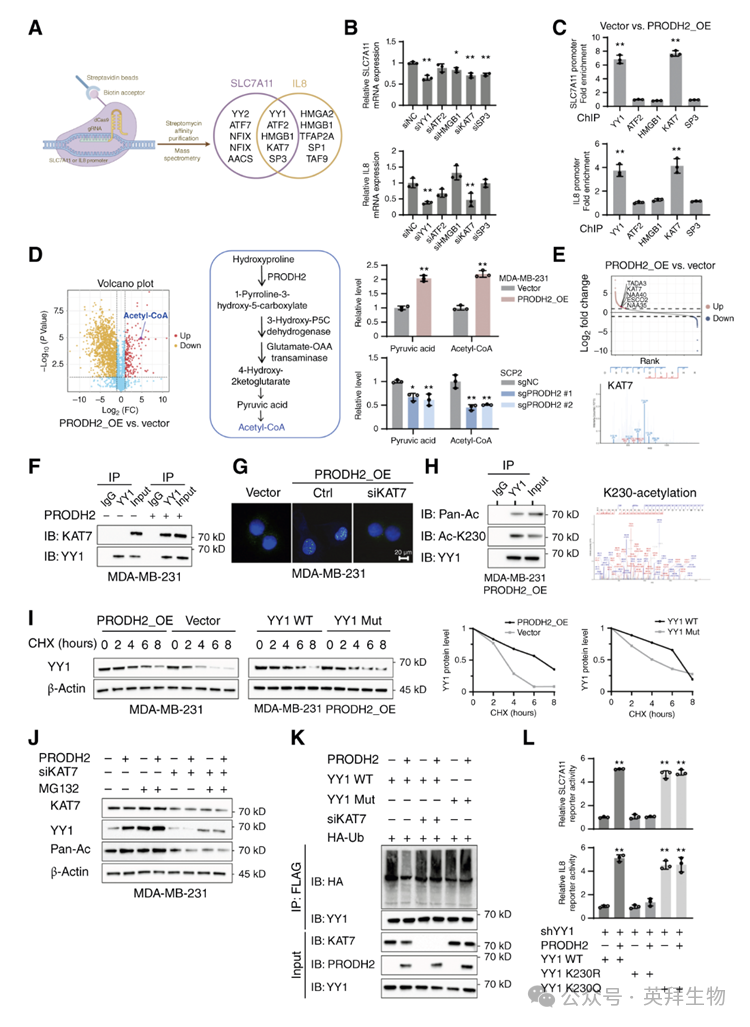
图5:PRODH2介导的Hyp通过增强YY1乙酰化促进骨转移
6、药物抑制 PRODH2 阻断乳腺癌骨转移
最后,作者评估了 PRODH2 抑制剂(N-丙炔甘氨酸)的干预效果。体外实验显示,该抑制剂显著抑制破骨细胞分化(图 6A)。体内实验进一步证实,其可显著减少骨转移灶、破骨细胞生成及胶原降解(图 6B)。动物模型 IHC 显示,PRODH2 敲除降低 YY1 核定位及 SLC7A11、IL8 表达(图 6C)。临床骨转移标本分析亦验证 PRODH2–SLC7A11–IL8 轴的相关性(图 6D)。
综上所述,PRODH2 通过抑制铁死亡、刺激破骨细胞分化,加速胶原降解,从而促进乳腺癌骨转移(图 6E),为其靶向治疗提供了理论依据。
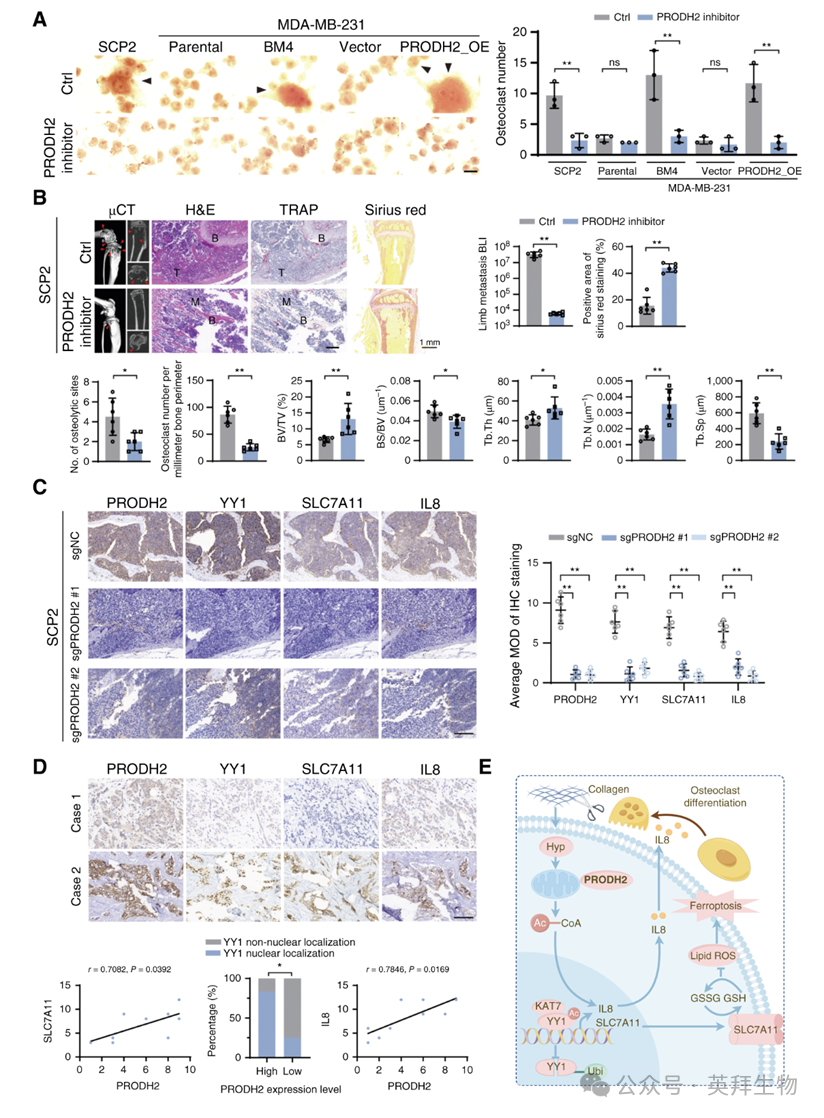
图6:体内药物抑制PRODH2可阻断乳腺癌细胞骨转移
结论:
总体而言,作者的研究证实,羟脯氨酸(Hyp)代谢重编程通过一种全新的PRODH2介导机制,成为乳腺癌骨转移的关键调控环节。具体而言,Hyp来源的乙酰辅酶A促进KAT7依赖的YY1-K230位点乙酰化,进而协同上调SLC7A11(赋予铁死亡抵抗)和IL8(促进破骨细胞生成),形成维持转移进展的正反馈环路。该发现不仅揭示了骨转移微环境中此前未被认识的代谢-表观遗传交叉对话,也为治疗骨转移性乳腺癌提供了可干预的新靶点。
参考文献:
Gong H, Li Y, Yang W, Xie Z, Hu J, Li P, Xu R, Li Y, Tao T, Li R, Liu S, Zhu Y, Song L, Fang L. PRODH2-Mediated Metabolism in the Bone Microenvironment Promotes Breast Cancer Metastasis. Cancer Res. 2025 Aug 1. doi: 10.1158/0008-5472.CAN-24-4391. Epub ahead of print. PMID: 40749014.