






乳酸积累诱导H4K12乳酸化修饰 通过激活超级增强子驱动的RAD23A表达促进卵巢癌尼拉帕尼耐药
卵巢癌是复发率和死亡率最高的妇科恶性肿瘤。尽管尼拉帕利能有效延缓其进展,但耐药性难题依然存在。本研究通过构建尼拉帕利耐药卵巢癌细胞系,利用RNA原位构象测序技术鉴定出异常活化的增强子及其相关靶基因。值得注意的是,靶基因RAD23A在耐药细胞中显著上调,抑制RAD23A可恢复细胞敏感性。此外,耐药细胞中糖酵解异常活化诱导乳酸积累,进而促进组蛋白H4K12赖氨酸残基的乳酸化修饰。相关性分析显示,在卵巢癌中丙酮酸激酶M和乳酸脱氢酶A等关键糖酵解酶与RAD23A表达呈显著正相关。研究进一步发现H4K12la通过MYC转录因子激活尼拉帕利耐药相关超级增强子及RAD23A表达,从而增强DNA损伤修复能力并促进卵巢癌细胞的耐药性。综上,本研究表明乳酸积累导致组蛋白H4K12la乳酸化修饰,进而通过超级增强子上调RAD23A异常表达,最终促进卵巢癌细胞对尼拉帕利的耐药,提示RAD23A可作为尼拉帕利耐药卵巢癌的潜在治疗靶点。该研究于2025年5月发表在《Nature Communications》,IF:15.8。
技术路线:
主要研究结果:
1.基于RIC-seq技术构建的尼拉帕尼耐药卵巢癌细胞中超级增强子与靶基因启动子互作图谱
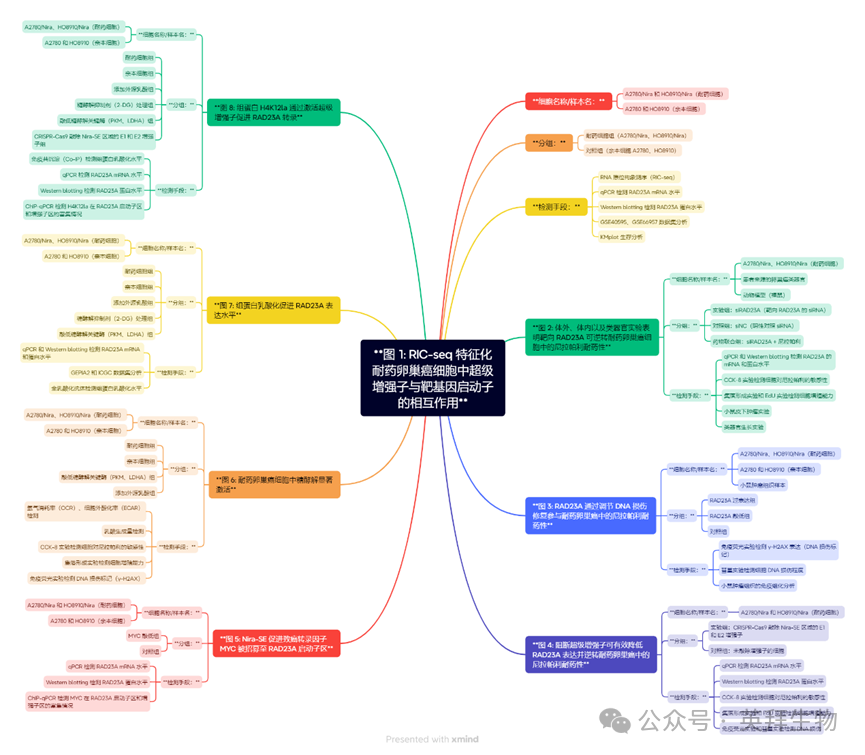
为获取卵巢癌尼拉帕尼耐药相关超级增强子(SEs)与靶基因的互作图谱,本研究通过以下方法构建耐药细胞模型:首先使用卡铂(10 µM)处理A2780和HO8910细胞48小时模拟药物诱导的DNA损伤,继而采用浓度梯度法诱导尼拉帕尼耐药(图1A)。CCK-8实验检测结果显示尼拉帕尼的IC50值显著升高(图1B-E)。通过整合A2780/Nira和HO8910/Nira细胞的RIC-seq数据与公共增强子数据库,最终获得与尼拉帕尼耐药相关的超级增强子-靶基因互作图谱(图1F,附表S6)。RIC-seq测序分析发现,位于chr19:13004808-13072187区段的超级增强子(命名为Nira-SE)与多个靶基因启动子存在频繁互作,包括RAD23A、RNU2-2P、WDR74和RP11-424C20.2。其中,Nira-SE与RAD23A基因启动子区域的互作信号最为显著(图1G)。
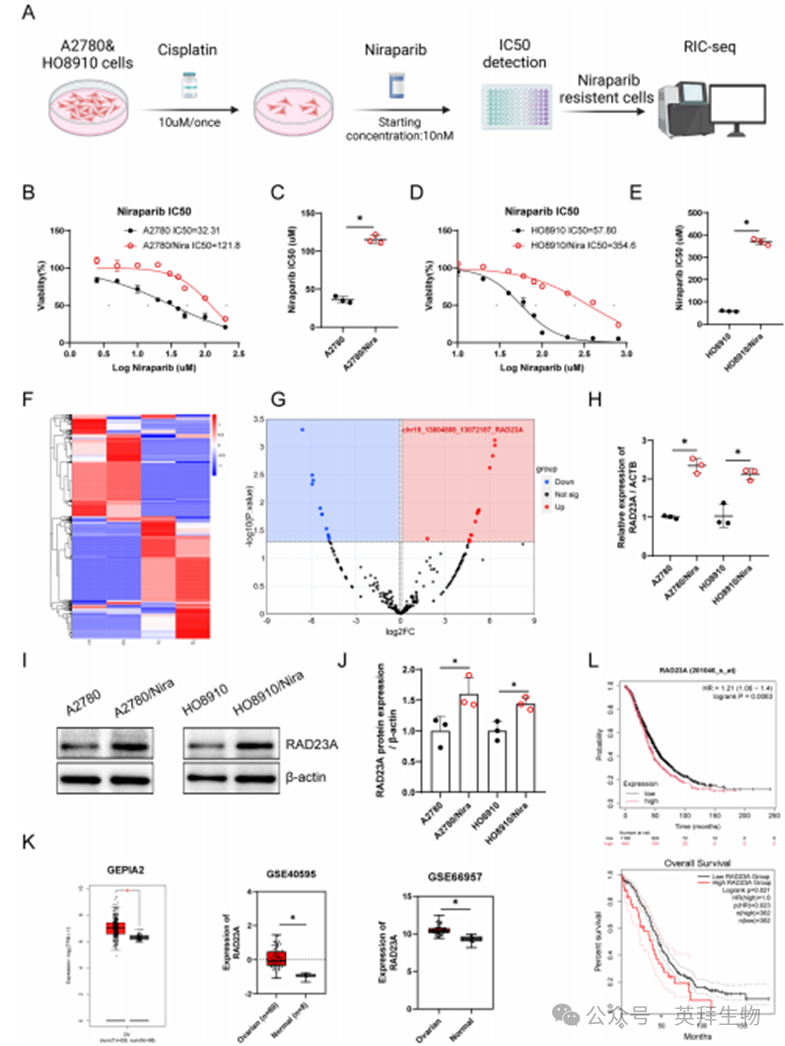
图1.基于RIC-seq技术构建的尼拉帕尼耐药卵巢癌细胞中超级增强子与靶基因启动子互作图谱
2.体外、体内及类器官实验表明靶向RAD23A可逆转卵巢癌尼拉帕尼耐药
为阐明RAD23A在卵巢癌尼拉帕尼耐药中的作用,本研究检测了卵巢癌细胞系A2780、A2780/Nira、HO8910和HO8910/Nira中RAD23A的表达水平。值得注意的是,尼拉帕尼耐药细胞中RAD23A的信使RNA(mRNA)和蛋白表达量均显著高于非耐药细胞(图1H-J)。通过对GSE40595、GSE66957数据集及GEPIA2数据库[19,20]的分析发现,RAD23A在卵巢癌组织中表达上调(图1K);KMplot[21]生存分析和GEPIA2数据集显示,RAD23A高表达与卵巢癌患者不良预后相关(图1L)。
为评估RAD23A在尼拉帕尼耐药卵巢癌细胞中的功能,我们通过转染siRAD23A敲低其表达(图2A-B)。CCK-8实验表明,敲低RAD23A可增强卵巢癌细胞对尼拉帕尼的敏感性(图2C-D)。此外,克隆形成实验和EdU检测显示,敲低RAD23A后耐药细胞的增殖能力显著降低(图2E-F)。值得注意的是,在铂类药物预处理的A2780和HO8910细胞中,过表达RAD23A可增强尼拉帕尼耐药性并提升细胞增殖能力(图2G-J)。
构建尼拉帕尼耐药细胞移植瘤模型及卵巢癌患者来源类器官模型以验证RAD23A的体内治疗效果。值得注意的是,与siNC组相比,siRAD23A组皮下肿瘤形成显著受抑;siRAD23A联合尼拉帕尼治疗组较siNC联合尼拉帕尼组表现出更强的肿瘤生长抑制效果(图2K)。类似地,类器官分析显示:与siNC组相比,siRAD23A组类器官生长明显受抑;且siRAD23A联合尼拉帕尼组较siNC联合尼拉帕尼组对类器官生长的抑制作用更为显著(图2L-M)。
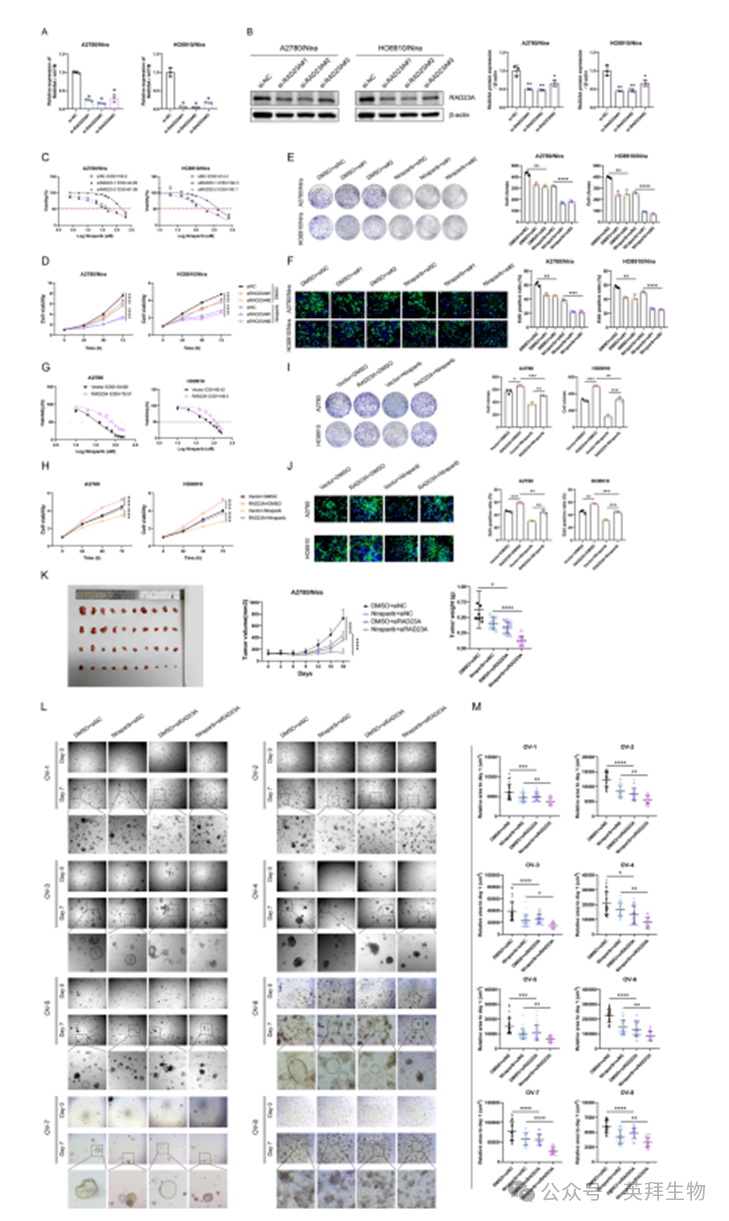
图2.体外、体内及类器官实验表明靶向RAD23A可逆转卵巢癌尼拉帕尼耐药
3.RAD23A通过调控DNA损伤修复参与卵巢癌尼拉帕尼耐药机制
作为核苷酸切除修复(NER)通路的重要组分,RAD23A能够修复由紫外线辐射和化学药物等多种因素引起的DNA损伤。为探究其在尼拉帕尼耐药细胞中的功能,我们评估了RAD23A对DNA损伤修复的影响。免疫荧光实验显示,在铂类预处理的A2780和HO8910细胞中,过表达RAD23A可降低双链DNA损伤标志物γ-H2AX的表达水平(图3A-B)。彗星实验进一步证实,RAD23A过表达后DNA损伤程度减轻,表明高水平RAD23A能增强DNA损伤修复能力(图3C-D)。此外,下调RAD23A则显著加剧DNA损伤(图3E-H)。小鼠移植瘤组织的免疫组化结果同样显示,siRAD23A联合尼拉帕尼治疗组中γ-H2AX表达显著升高(图3I)。这些结果共同表明,RAD23A通过调控DNA损伤修复参与卵巢癌尼拉帕尼耐药机制。
图3.RAD23A通过调控DNA损伤修复参与卵巢癌尼拉帕尼耐药机制
4.Nira-SE调控靶基因RAD23A的表达机制研究
为阐明RAD23A在卵巢癌化疗耐药中表观遗传重塑的机制,我们进行了生物信息学分析。通过FANTOM5数据库[22]在Nira-SE区域鉴定出两个增强子:chr19:13023589-13024232(命名为E1)和chr19:13048278-13048689(命名为E2)(图4A)。随后利用CRISPR-Cas9基因编辑技术分别敲除E1和E2增强子(图4B-C),以评估其对RAD23A转录活性的影响。qPCR和蛋白质印迹实验结果显示,单独敲除E1或E2均能显著降低RAD23A表达,而同时敲除两个增强子则使RAD23A表达进一步下调(图4D-E)。为评估阻断Nira-SE对卵巢癌尼拉帕尼耐药的影响,本研究在A2780/Nira和HO8910/Nira细胞中分别敲除E1和E2增强子。CCK-8实验表明,敲除E1或E2均可增强耐药细胞对尼拉帕尼的敏感性(图4F-G)。进一步通过克隆形成实验与EdU检测发现,敲除E1或E2后耐药细胞的增殖能力显著下降(图4H-I)。此外,免疫荧光实验显示增强子敲除后γ-H2AX表达显著升高(图4J);彗星实验亦证实DNA损伤程度加剧(图4K)。
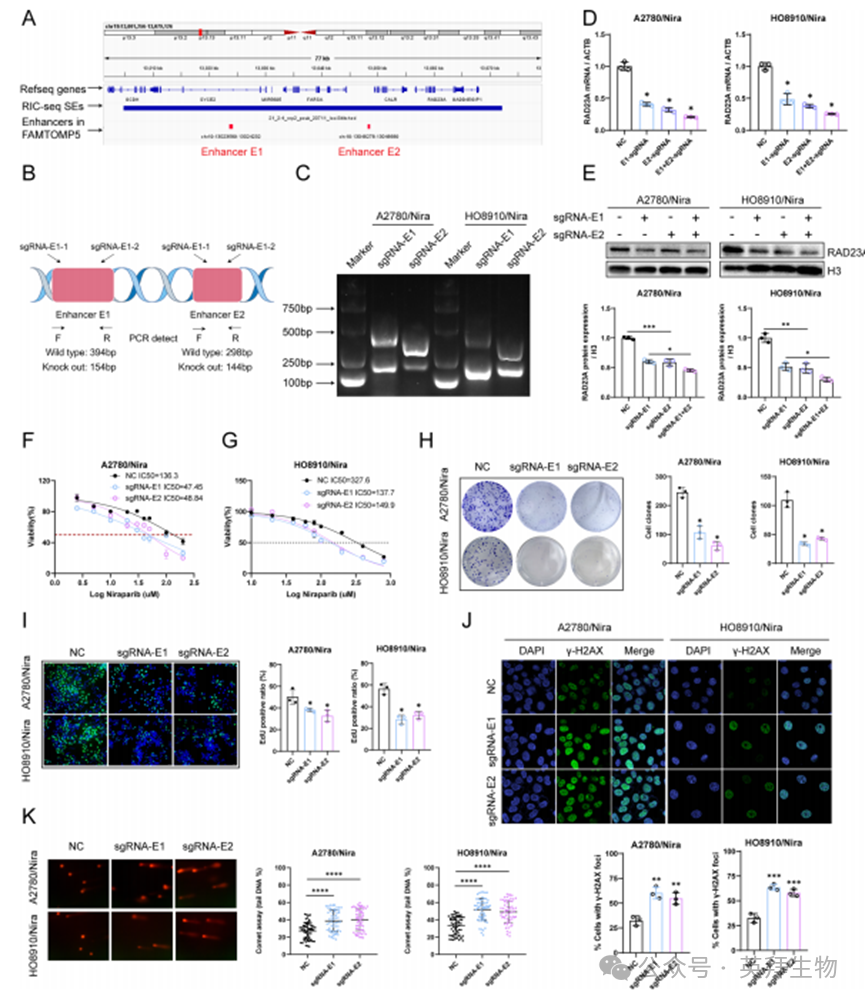
图4. Nira-SE调控靶基因RAD23A的表达机制研究
5.Nira-SE通过促进致癌转录因子MYC在启动子区的富集调控转录
转录因子(TFs)介导超级增强子调控网络并以基因特异性方式启动转录。本研究通过整合JASPAR、PROMO、Cistrome和ENCODE四个数据库的预测结果,鉴定出7个与RAD23A相关的转录因子:YY1、USF2、MYC、E2F1、RELA、GATA1和USF1(图5A)。后续功能验证发现,敲低MYC可显著降低RAD23A的mRNA(图5B-C)和蛋白表达水平(图5D-F)。ChIP-qPCR分析显示,在A2780和HO8910细胞中,MYC富集于RAD23A的启动子区(图5G-H)以及增强子E1和E2区域(图5I-J),且在其相应尼拉帕尼耐药细胞中的富集程度更为显著(图5K-M),表明MYC是调控耐药卵巢癌细胞中RAD23A转录的关键因子。进一步评估超级增强子对MYC介导的RAD23A转录激活的影响,发现敲除E1和E2后,MYC在RAD23A启动子区的富集显著减少(图5N),表明增强子缺失有效抑制了MYC的启动子富集,从而影响其转录活性。
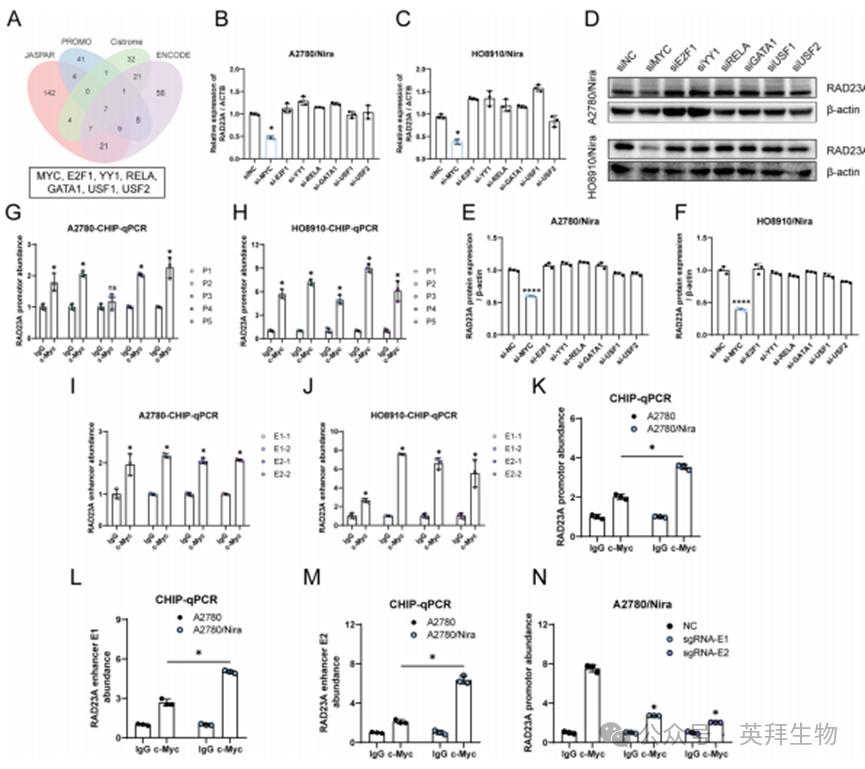
图5. Nira-SE通过促进致癌转录因子MYC在启动子区的富集调控转录
6.尼拉帕尼耐药卵巢癌细胞中糖酵解显著激活
糖酵解可驱动肿瘤细胞的恶性进展及耐药性[23]。本研究旨在探讨糖酵解及组蛋白乳酸化修饰是否参与卵巢癌尼拉帕尼耐药过程中RAD23A超级增强子(SE)的形成与激活。通过检测耐药细胞的糖酵解相关指标发现:与亲本细胞相比,A2780/Nira和HO8910/Nira细胞的耗氧率(OCR)显著降低(图6A),而细胞外酸化率(ECAR)升高(图6B),乳酸生成量明显增加(图6C)。
为探究乳酸积累机制,我们检测了丙酮酸脱氢酶复合体关键限速酶PDHA1的表达与修饰状态。结果显示,虽然PDHA1总蛋白水平无变化,但其Ser232和Ser300位点的磷酸化水平在耐药细胞中上调(附图1A)。已有研究证实这两个位点的磷酸化会抑制PDHA1活性并介导乳酸过量生成[26]。同时,我们检测了维持肿瘤细胞乳酸稳态的主要转运蛋白MCT1/4,发现其蛋白水平(附图1B)及细胞膜定位(附图1C)在耐药细胞中均未发生改变。这些结果表明尼拉帕尼耐药卵巢癌细胞中糖酵解被激活并导致乳酸积累。
体外实验进一步证实,敲低糖酵解关键酶PKM或LDHA可显著增强耐药细胞对尼拉帕尼的敏感性(图6D),抑制细胞增殖(图6E),并提升DNA损伤水平(图6F)。相反,添加外源性乳酸则促进卵巢癌细胞增殖及尼拉帕尼耐药,同时降低DNA损伤程度。
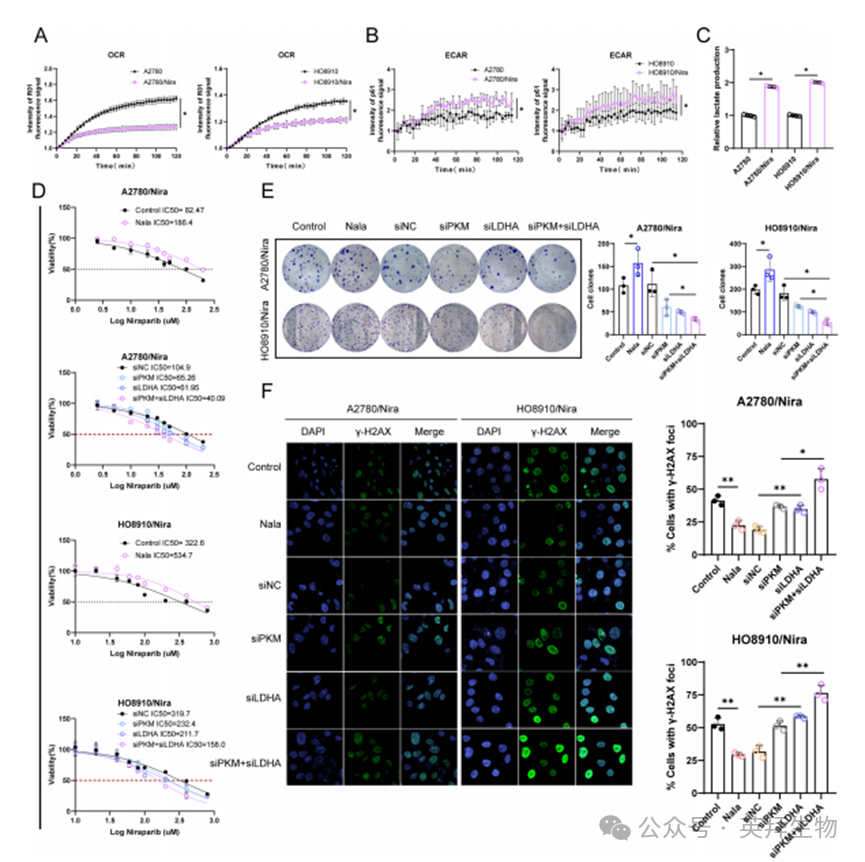
图6. 尼拉帕尼耐药卵巢癌细胞中糖酵解显著激活
7.组蛋白乳酸化修饰促进RAD23A表达
尽管耐药细胞中糖酵解激活导致乳酸积累,但其对RAD23A基因表达的影响尚不明确。通过分析GEPIA2和ICGC数据库中己糖激酶2、PKM、LDHA与RAD23A的表达相关性,发现PKM、LDHA与RAD23A表达呈显著正相关(图7A-B)。此外,PKM与LDHA在卵巢癌组织中的表达均显著上调(图7C)。为阐明糖酵解通路通过组蛋白乳酸化对RAD23A转录表达的调控作用,我们采用泛乳酸化抗体检测整体乳酸化水平。结果显示,尼拉帕尼耐药细胞中组蛋白乳酸化水平显著升高(图7D);添加外源性乳酸(乳酸钠,Nala)后,组蛋白乳酸化水平与RAD23A表达均进一步上调(图7E-F)。相反,当使用糖酵解抑制剂2-DG处理卵巢癌细胞时,组蛋白乳酸化与RAD23A表达均显著降低(图7G)。在耐药细胞中敲低PKM或LDHA可明显降低RAD23A的mRNA和蛋白水平,而补充外源性乳酸则能恢复RAD23A表达(图7H-I)。
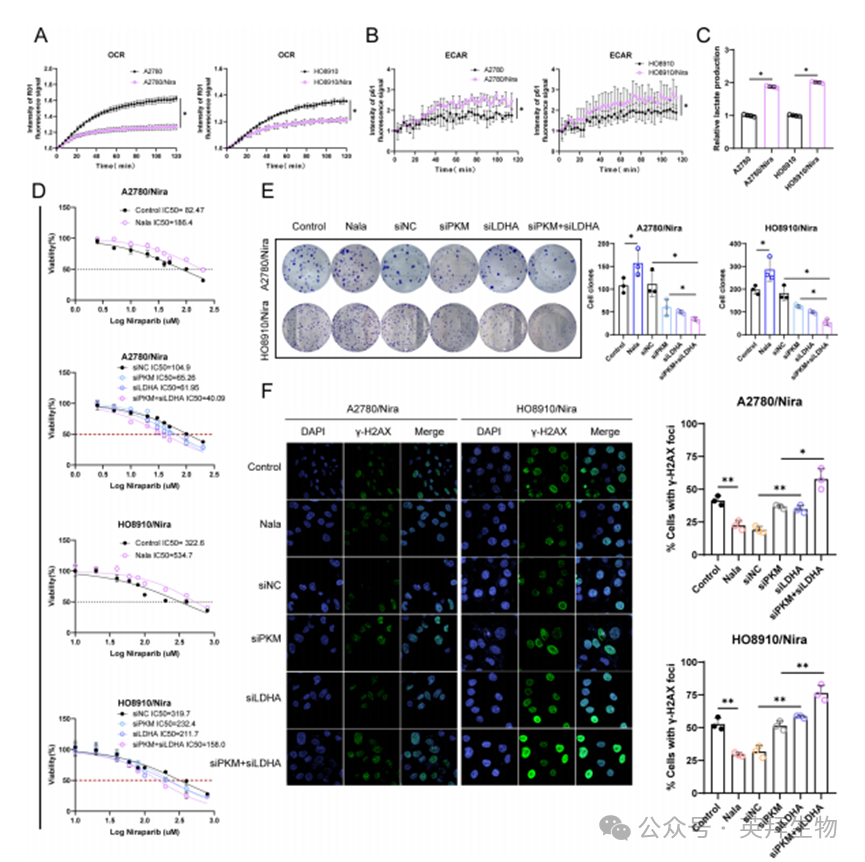
图7. 组蛋白乳酸化修饰促进RAD23A表达
8.H4K12乳酸化通过激活Nira-SE促进RAD23A转录
为验证组蛋白修饰位点在转录调控中的作用,我们检测了尼拉帕尼耐药细胞中四种核心组蛋白(H2A、H2B、H3和H4)的乳酸化水平。免疫共沉淀(CO-IP)结果显示,耐药细胞中H4组蛋白的乳酸化修饰显著增强(图8A-B)。进一步对H4蛋白常见乳酸化位点进行筛选,发现包括H4K5la、H4K8la、H4K12la和H4K16la在内的多个位点中,H4K12la在耐药细胞中的上调最为显著(图8C-D)。外源性乳酸与糖酵解抑制剂2-DG可分别显著上调和下调H4K12la水平(图8E-F)。此外,敲低PKM或LDHA会导致H4K12la修饰降低,而补充外源性乳酸则能逆转该效应(图8G-H)。这些结果表明H4K12乳酸化是调控卵巢癌尼拉帕尼耐药的关键组蛋白修饰位点。
为深入验证H4K12la对靶基因RAD23A的转录调控机制,我们检测了H4K12la在RAD23A基因启动子区及增强子E1/E2区域的富集情况。ChIP实验结果显示,在非耐药亲本细胞中即可检测到H4K12la在RAD23A基因启动子、E1和E2区域的富集(图8I-J),而在尼拉帕尼耐药细胞中,这些区域的H4K12la富集水平进一步升高(图8K-L)。机制探索实验表明:敲低PKM/LDHA可同时降低MYC和H4K12la在RAD23A启动子/增强子区的富集(附图2);在A2780和HO8910细胞中添加外源性乳酸能增强MYC在RAD23A启动子区的富集,而在耐药细胞中使用2-DG则产生抑制作用(附图3)。值得注意的是,敲低MYC并不影响H4K12la在RAD23A调控区域的富集,且H4K12la与MYC蛋白无直接相互作用(附图4)。最终,通过CRISPR-Cas9技术敲除Nira-SE中的E1/E2区域后,RAD23A启动子区的H4K12la富集显著降低,且该效应无法通过外源性乳酸补充逆转(图8M)。这些结果证实Nira-SE通过其空间结构功能促进H4K12la乳酸化修饰在靶基因启动子区的富集,从而激活RAD23A的转录表达。
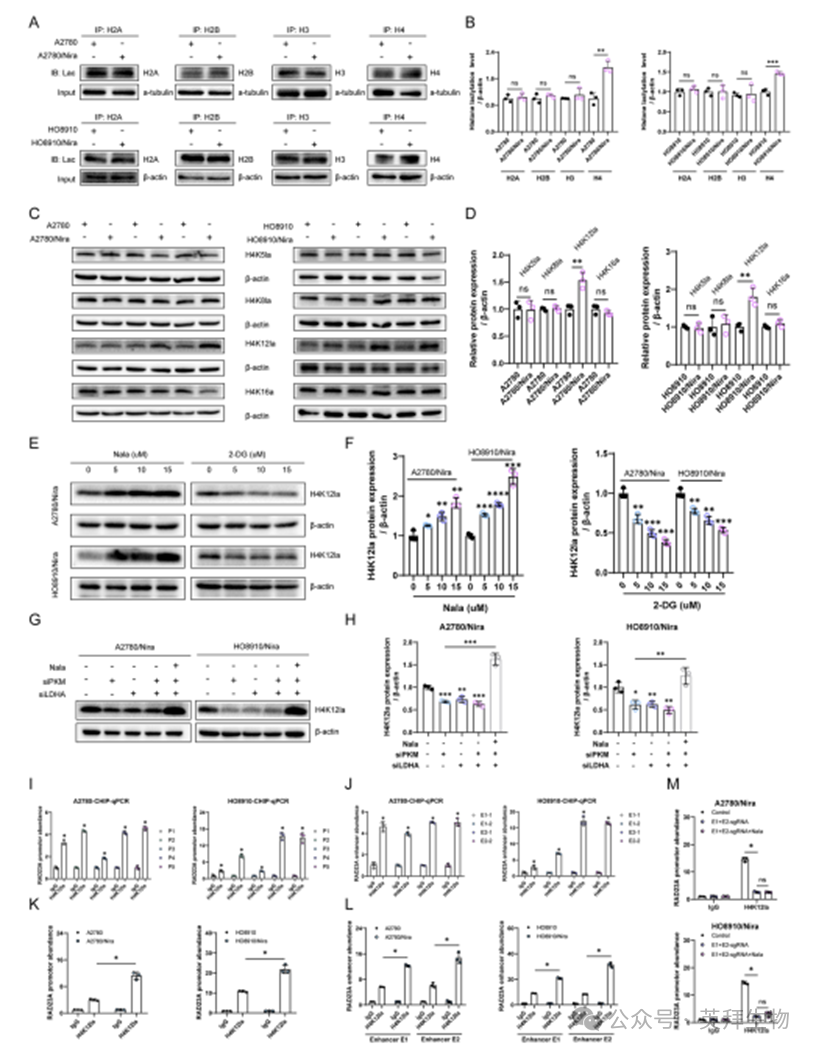
图8.H4K12乳酸化通过激活Nira-SE促进RAD23A转录
结论:
本研究揭示了在卵巢癌中,药物耐药性与糖酵解激活、组蛋白乳酸化修饰、超级增强子(SE)以及DNA损伤修复基因RAD23A的异常激活密切相关。研究发现,耐药细胞中糖酵解的增强导致乳酸积累,进而增加组蛋白H4K12的乳酸化修饰(H4K12la),并通过激活位于RAD23A基因上游的超级增强子(Nira-SE),招募致癌转录因子MYC,从而促进RAD23A基因的转录和表达,增强DNA损伤修复能力,导致对尼拉帕利(niraparib)的耐药性。通过靶向RAD23A或阻断Nira-SE,可以恢复卵巢癌细胞对尼拉帕利的敏感性,为克服卵巢癌耐药性提供了新的治疗靶点。
参考文献:
Lu B, Chen S, Guan X, Chen X, Du Y, Yuan J, Wang J, Wu Q, Zhou L, Huang X, Zhao Y. Lactate accumulation induces H4K12la to activate super-enhancer-driven RAD23A expression and promote niraparib resistance in ovarian cancer. Mol Cancer. 2025 Mar 19;24(1):83.