






抑癌基因SLC9A2通过抑制血管生成阻断结直肠癌转移并逆转免疫治疗耐药
结直肠癌(CRC)是一种常见且侵袭性强的恶性肿瘤,常转移至肝脏,治疗面临巨大挑战。尽管其临床意义重大,CRC肝转移及免疫治疗耐药的机制仍不清楚。本研究采用新策略——构建高转移CRC细胞系——探讨驱动CRC转移的分子机制。为明确特定基因在CRC肝转移中的作用,作者对原代CRC细胞持续施加选择压力,建立两株高转移细胞系(LoVo-Hm与HCT116-Hm)。RNA测序筛选转移细胞中差异表达基因,并通过迁移、侵袭、血管生成实验及ELISA检测VEGFA分泌,验证SLC9A2功能。结果在人CRC组织及公共数据集中进一步验证,以评估其临床意义。高转移CRC细胞系中SLC9A2显著下调。机制上,SLC9A2通过抑制STAT3信号通路阻断上皮-间质转化(EMT)及转移;同时减少VEGFA分泌,使肿瘤血管正常化并重塑肿瘤微环境(TME),最终增强抗肿瘤免疫。CRC组织分析显示,肿瘤组织SLC9A2表达低于邻近正常组织,且与TNM分期呈负相关。重要的是,免疫治疗队列中SLC9A2高表达者治疗反应更佳。SLC9A2通过调控STAT3通路及肿瘤血管,在CRC转移、血管生成及TME重塑中发挥关键作用,可作为潜在预后生物标志物和治疗靶点。靶向SLC9A2有望增强免疫应答、改善CRC治疗效果,为未来治疗策略提供新方向。该文章于2025年6月发表在《JOURNAL OF EXPERIMENTAL & CLINICAL CANCER RESEARCH》,IF:12.8。
技术路线:

主要研究结果:
1.高转移性CRC模型构建及转移驱动基因探索
肿瘤异质性体现在增殖、转移、耐药等多个方面。为研究这些特征,作者选取源自原发CRC的HCT116细胞系和来自淋巴结转移的LoVo细胞系作为基础,施加体内外持续选择压力,成功构建高转移株HCT116-Hm与LoVo-Hm(图1A)。与亲本细胞相比,筛选获得的细胞迁移和侵袭能力显著增强(图1B-C)。腹腔播散及脾-肝转移模型进一步证实该筛选体系可有效获得高转移能力细胞(图1D-E)。对LoVo及其高转移衍生物LoVo-Hm进行转录组测序,比较表达谱变化(图1F);同时分析包含原发CRC及配对肝转移数据的GSE14297数据集(图1G)。首先过滤FPKM>0的基因,再以|log₂FC|>2、FDR<0.05为标准筛选差异基因,取上调及下调前20位。将该结果与GSE14297中原发瘤与肝转移瘤的差异基因交集,仅SLC9A2同时出现,遂确定其为后续研究对象(图1H)。值得注意的是,SLC9A2在LoVo-Hm中显著下调,在肝转移组织中也低于原发瘤。

图1:结直肠癌肝转移中SLC9A2表达下调
利用GSE146771单细胞数据进一步验证SLC9A2表达是否局限于肿瘤细胞。结果显示,该基因仅在恶性上皮细胞中表达,基质及免疫细胞未见信号(图1I)。GSE178341数据集亦证实SLC9A2表达局限于肿瘤上皮成分(图1J-M)。再次分析GSE14297发现,原发瘤(PT)SLC9A2水平显著低于肝转移(LM)(图1N)。构建原发CRC与肝转移配对类器官(图1O),H&E染色显示类器官具有核质比异常等典型肿瘤特征,病理检测证实其表达CRC标志(图1Q)。提取3例患者来源类器官蛋白,定量显示肝转移类器官SLC9A2蛋白水平显著低于原发瘤类器官(图1P)。LoVo与LoVo-Hm细胞中SLC9A2 mRNA及蛋白检测均提示高转移株表达显著降低(图1R)。TCGA数据库Kaplan-Meier分析显示,SLC9A2 mRNA水平与CRC患者总生存呈正相关。
2.SLC9A2低表达与结直肠癌患者TNM分期晚期及预后不良相关
SLC9A2在转移灶中显著下调,并与CRC患者不良预后密切相关。为明确其与临床特征的关系,作者整合GEO与TCGA公共数据集进行分析。结果显示,SLC9A2表达水平是CRC患者总生存(OS)、无复发生存(RFS)、疾病特异生存(DSS)及无进展生存(PFS)的有利预测因子(图2A)。利用TCGA及三个GEO数据集检测CRC标本中SLC9A2 mRNA表达,结果一致表明肿瘤组织表达显著低于邻近正常组织(图2B)。此外,微卫星高度不稳定(MSI-H)CRC标本的SLC9A2表达高于微卫星稳定(MSS)标本(图2C)。值得注意的是,TNM晚期患者SLC9A2转录水平显著降低(图2D),且其表达随T、N、M分期进展呈阶梯式下降(图2E-G)。通过Timer数据库分析SLC9A2表达与免疫细胞浸润的关系,发现其与肿瘤相关成纤维细胞及M2型巨噬细胞呈负相关,而与CD8⁺ T细胞呈正相关(图2H)。进一步评估SLC9A2对免疫治疗的影响,结果显示其与LAG3、CTLA4、CD274、TGFB1、TIGIT、ADORA2A、CSF1R、KDR等8种免疫检查点分子均呈负相关(图2I),提示SLC9A2可能增强免疫治疗疗效。为验证上述关联,作者分析CRC免疫治疗单细胞数据集GSE178341,发现基线组织高表达SLC9A2的患者对免疫治疗反应良好,而低表达者均无治疗响应(图2J-M)。
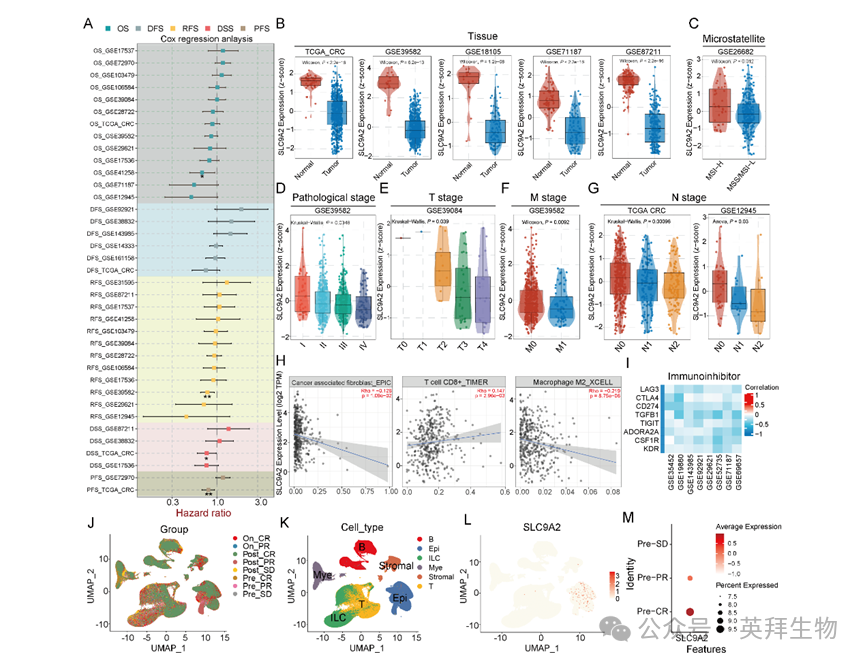
图2:SLC9A2低表达与不良临床特征相关并伴随免疫抑制微环境
3.SLC9A2体外抑制CRC细胞恶性进展
为验证SLC9A2在CRC中的抑癌功能,作者采用过表达质粒及siRNA分别上调和下调其表达,RT-qPCR证实转染效率(Figure S1)。鉴于SLC9A2源自转移模型,重点考察其对迁移与侵袭的影响。Transwell实验显示,敲低SLC9A2后,穿膜细胞数显著减少,Matrigel侵袭能力亦降低(图3A-B)。反之,在LoVo-Hm细胞中恢复SLC9A2表达,穿膜及侵袭细胞数均下降(图3C)。划痕实验进一步证实:siRNA组LoVo细胞迁移距离增加(图3D),而过表达SLC9A2的LoVo-Hm细胞迁移距离缩短(图3E)。上述结果提示SLC9A2抑制CRC细胞迁移与侵袭。
EMT是肿瘤转移的关键环节,与迁移、侵袭密切相关。GSEA分析显示,SLC9A2低表达组EMT信号显著富集(图3F);且SLC9A2与上皮标志物CDH1正相关,与间质标志物CDH2、SNAIL1负相关(图3F),提示其抑制EMT进程。Western blot进一步验证:敲低SLC9A2后,间质标志物N-cadherin、Snail上调,上皮标志物E-cadherin、Claudin-1下调;过表达SLC9A2则呈现相反趋势(图3G-H)。综上,SLC9A2通过抑制EMT阻断CRC细胞迁移与侵袭。
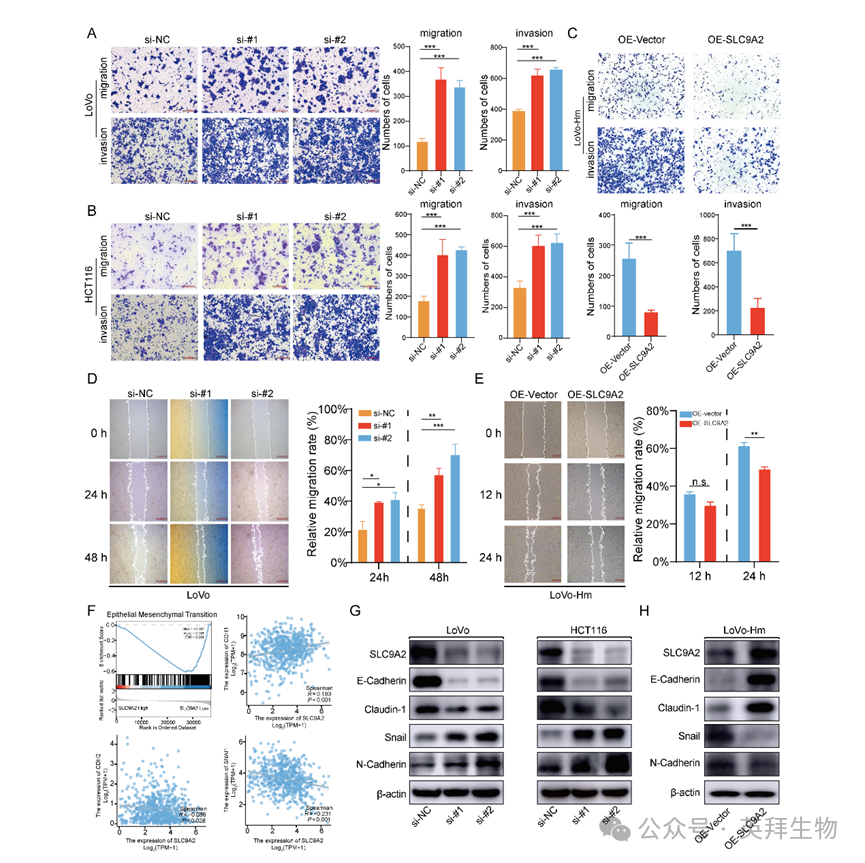
图3:SLC9A2抑制CRC细胞迁移侵袭并逆转EMT进程
4.SLC9A2体内抑制结直肠癌肝转移
为验证SLC9A2的体内功能,作者采用CRC脾-肝转移模型评估其对肿瘤细胞迁移和侵袭的影响。结果显示,与对照组相比,SLC9A2过表达组肝脏转移结节数量显著减少(图4A-E)。肝组织H&E染色亦显示,SLC9A2过表达组转移灶明显减少(图4F)。免疫组化(IHC)结果显示,SLC9A2过表达显著上调上皮标志物E-cadherin表达,下调间质标志物N-cadherin和Snail表达。此外,作者检测了肝转移组织中血管生成标志物CD31的表达水平,发现SLC9A2过表达组肿瘤组织中CD31表达显著降低,提示SLC9A2可能参与肿瘤血管生成过程(图4G)。进一步,作者对同一患者的原发CRC病灶及对应肝转移灶进行IHC染色,发现SLC9A2在原发肿瘤中的表达高于肝转移灶,而CD31在肝转移灶中的表达高于原发灶。该结果进一步支持SLC9A2与CD31之间的负相关关系,提示SLC9A2在肿瘤血管生成中发挥作用(图4H)。此外,作者收集肝转移灶标本进行RT-qPCR和Western blot分析。如图4I-J所示,SLC9A2过表达在mRNA和蛋白水平均上调E-cadherin表达,下调N-cadherin和Vimentin表达。综上,SLC9A2在体内可抑制结直肠癌的血管生成和转移。
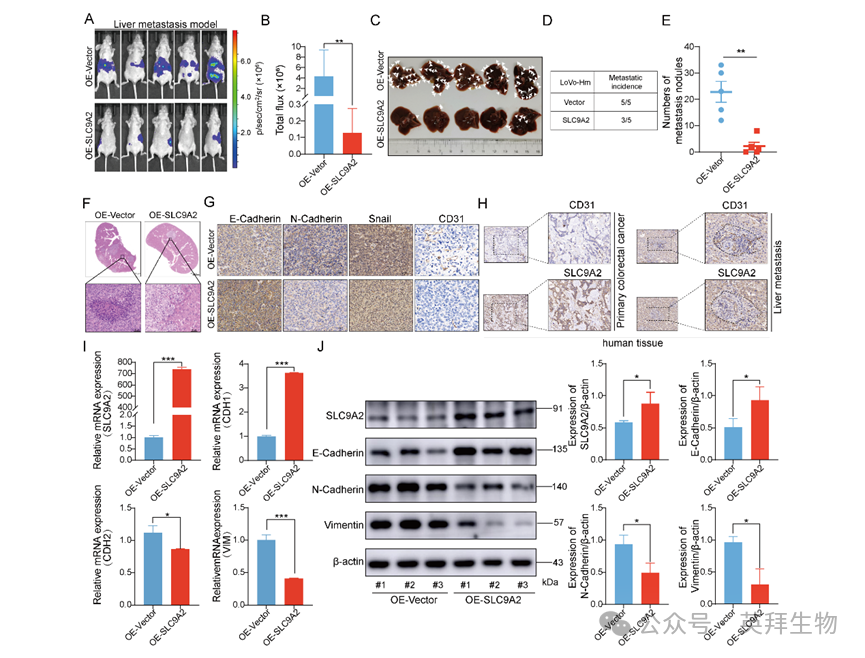
图4:SLC9A2在体内抑制结直肠癌肝转移
5.SLC9A2通过抑制JAK/STAT3信号通路阻断结直肠癌转移
随后,作者探讨SLC9A2抑制CRC转移的分子机制。对SLC9A2过表达及对照细胞进行RNA测序,KEGG分析显示差异基因富集于JAK/STAT信号通路(图5A)。鉴于JAK/STAT3通路激活参与肿瘤发生、进展、侵袭及转移,作者首先检测STAT3第705位酪氨酸磷酸化(p-STAT3^Y705^)水平。敲低SLC9A2后,HCT116与LoVo细胞中p-STAT3^Y705^表达升高(图5B);反之,在HCT116-Hm与LoVo-Hm中过表达SLC9A2则显著下调p-STAT3^Y705^(图5C)。为证实SLC9A2缺失主要通过促进p-STAT3^Y705^累积及其核转录激活发挥作用,作者采用STAT3磷酸化抑制剂Stattic。Stattic处理可完全阻断敲低SLC9A2所致的p-STAT3^Y705^升高(图5D)。核-质分离实验显示,si-SLC9A2组细胞核内p-STAT3^Y705^蛋白水平显著高于si-NC组,而Stattic可抑制该效应(图5E)。免疫荧光进一步验证,SLC9A2过表达减少p-STAT3核转位(图5F-G)。功能层面,敲低SLC9A2显著促进HCT116与LoVo细胞迁移侵袭,而Stattic处理可明显逆转该效应(图5H-I)。此外,作者构建同一患者原发灶与肝转移灶来源的类器官模型(图5J)。在原发灶类器官中敲低SLC9A2,肝转移灶来源类器官p-STAT3^Y705^表达升高;反之,在肝转移灶类器官中过表达SLC9A2则抑制p-STAT3^Y705^水平(图5K)。小鼠肝转移模型组织Western blot亦证实,体内过表达SLC9A2可降低p-STAT3^Y705^(图5L)。综上,SLC9A2通过抑制p-STAT3^Y705^发挥抗肿瘤作用。
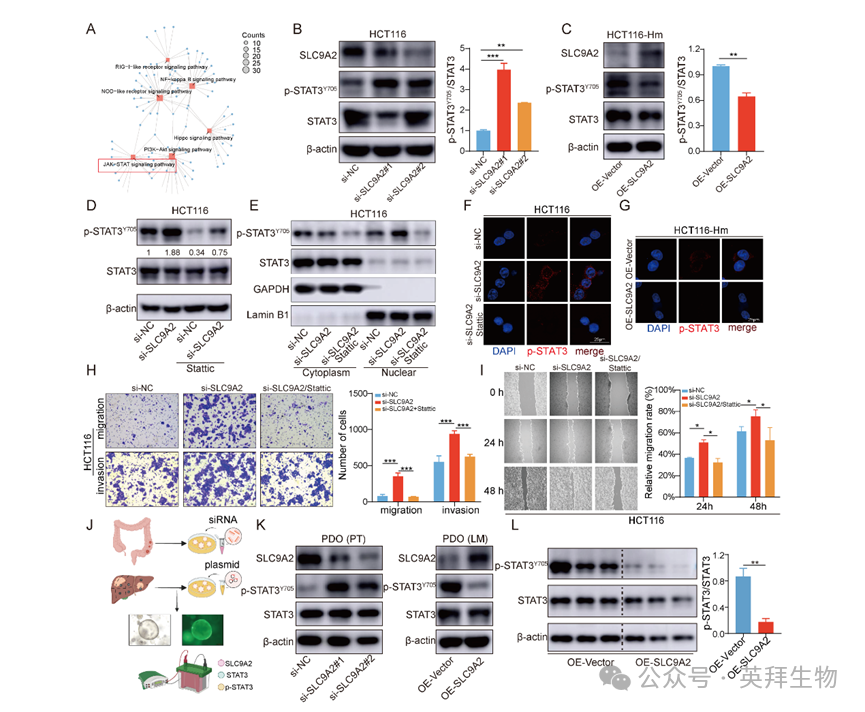
图5:SLC9A2通过抑制STAT3信号通路阻断CRC细胞迁移侵袭
6.SLC9A2通过抑制JAK/STAT3信号通路阻断结直肠癌转移
在高转移细胞中过表达SLC9A2可显著降低其体内肝转移能力。除通过抑制STAT3信号通路阻断肿瘤细胞迁移外,SLC9A2可能具备额外生物学功能以协同抑制CRC转移。差异基因GO分析显示,除JAK-STAT3通路外,VEGF信号通路亦显著富集,提示SLC9A2可能参与调控肿瘤血管生成(图6A)。血管生成是早期癌转移的关键环节,可促使肿瘤由休眠进入增殖期,加速生长与播散,故与CRC细胞的迁移侵袭能力密切相关。
利用CancerSEA单细胞平台解析CRC单细胞转录组数据,发现SLC9A2表达与肿瘤恶性表型呈负相关,尤其与血管生成呈显著负相关(图6B-C),提示其具有抗血管生成潜能。IHC染色亦显示,肝转移灶SLC9A2表达低于原发瘤,而血管标志物CD31在SLC9A2低表达区呈高表达,进一步支持其抑制血管生成的功能(图4H)。鉴于VEGFA在肿瘤血管生成中的核心作用,作者考察SLC9A2与VEGFA的关联。TNMplot数据库相关性分析显示二者表达呈负相关(图6D)。采用人脐静脉内皮细胞(HUVEC)进行成管实验,发现SLC9A2过表达肿瘤细胞条件培养基显著抑制HUVEC管腔形成;反之,敲低SLC9A2则促进成管。CRC类器官中敲低SLC9A2同样增强HUVEC成管能力(图6E-G)。内皮细胞迁移是血管生成的重要步骤。Transwell实验表明,SLC9A2过表达细胞条件培养基显著抑制HUVEC迁移与侵袭,而敲低SLC9A2的条件培养基则起促进作用(图6H-I)。GO分析提示差异基因亦富集于JAK-STAT3通路。作者前期已证实高转移细胞通过下调SLC9A2激活STAT3通路,促进迁移侵袭,由此推测STAT3亦可能参与血管生成。为验证此假设,在SLC9A2敲低细胞中加入STAT3磷酸化抑制剂Stattic,发现其可逆转由SLC9A2缺失诱导的VEGFA上调(图6J)。成管、迁移及EdU增殖实验进一步显示,Stattic可挽救SLC9A2敲低所致的HUVEC成管、迁移与增殖增强效应(图6K-L)。因此,SLC9A2通过抑制STAT3通路发挥抗血管生成作用。裸鼠Matrigel plug实验亦获得一致结果。H&E及CD31 IHC显示,与HCT116 si-NC组相比,si-SLC9A2共培养组胶栓内可见大量新生血管;而过表达SLC9A2则显著减少体内血管生成(图6M-N)。综上,SLC9A2在体外与体内均可显著抑制HUVEC血管生成能力。
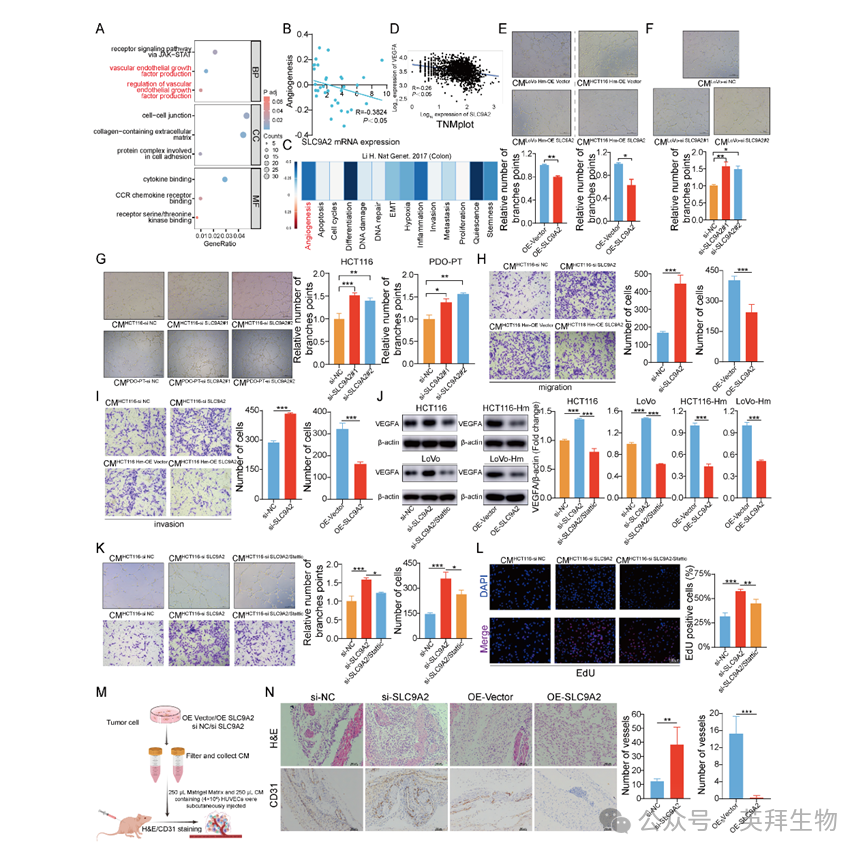
图6:SLC9A2抑制VEGFA表达并遏制肿瘤血管生成
7.SLC9A2通过抑制血管生成有效逆转结直肠癌免疫耐药
如图2所示,CRC免疫治疗队列中基线SLC9A2表达较高者治疗反应更佳。新近研究指出,抗血管生成治疗可增强效应T细胞浸润并促进血管正常化,同时减少调节性T细胞及髓源性抑制细胞招募[25]。据此,作者推测SLC9A2通过抑制STAT3通路阻断CRC转移与血管生成,进而协同增强免疫治疗疗效。为验证该假设,作者构建免疫治疗耐药亚系MC38-R:对MC38细胞长期低剂量诱导抗PD-1抗体,最终获得耐药株(图7A-B)。免疫治疗后皮下瘤IHC显示,MC38-R肿瘤内CD8⁺ T细胞浸润显著减少,血管密度明显增加,证实肿瘤血管生成通路激活参与免疫耐药过程(图7C)。采用原位过表达策略,于MC38-R移植瘤内注射Slc9a2过表达质粒(前期体外转染已验证表达,图7O)。皮下成瘤10天后,小鼠接受两周瘤内注射:Slc9a2过表达质粒+递送缓冲液、空载质粒+递送缓冲液,以贝伐珠单抗为阳性对照。单独抗PD-1治疗对MC38-R移植瘤无效;而Slc9a2质粒或贝伐珠单抗均显著增强抗PD-1的抑瘤效果(图7D-G)。ELISA证实,Slc9a2过表达组肿瘤VEGFA水平下降(图7H)。CD31 IHC显示,OE-Slc9a2+anti-PD-1组与贝伐珠单抗+anti-PD-1组血管数量显著少于其余各组(图7J)。肿瘤浸润免疫细胞分析表明,上述两组CD8⁺ TILs数量增加(图7K)。TUNEL检测显示,OE-Slc9a2+anti-PD-1与贝伐珠单抗+anti-PD-1组凋亡细胞增多,EdU染色示增殖降低(图7L-M)。ELISA进一步验证,OE-Slc9a2+anti-PD-1处理可促进颗粒酶B/IFN-γ分泌,尤以IFN-γ升高显著(图7N)。Western blot证实瘤内Slc9a2过表达效率,并显示p-STAT3与VEGFA表达下降(图7P)。综上,Slc9a2通过抑制STAT3通路降低VEGFA表达,从而逆转免疫治疗耐药。
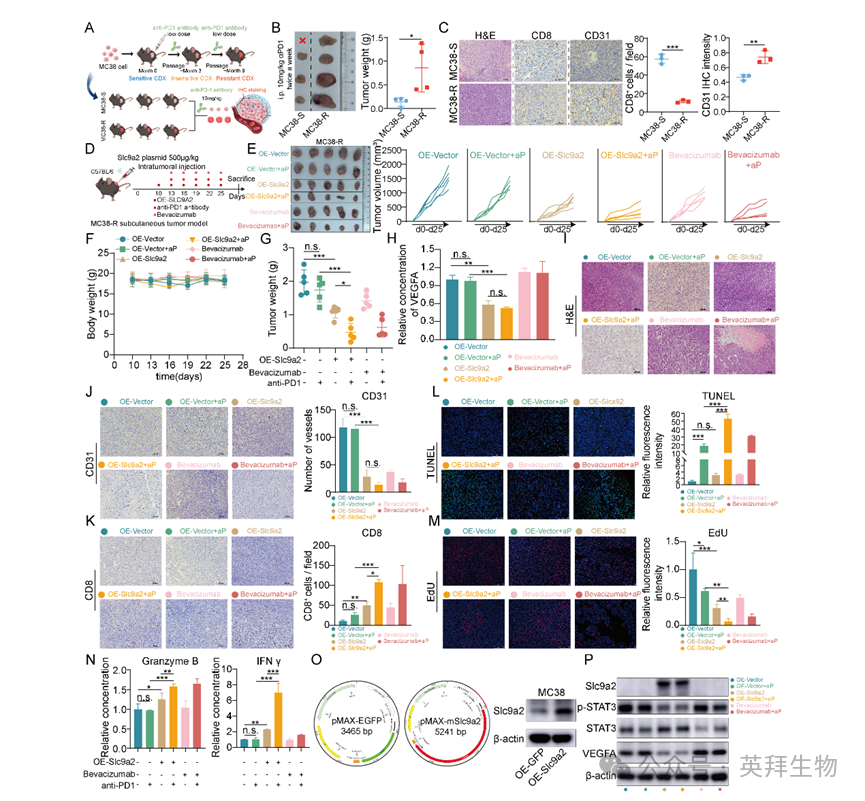
图7:Slc9a2通过抑制血管生成有效逆转结直肠癌免疫耐药
8.Ruxolitinib增强抗PD-1免疫治疗结直肠癌的疗效
目前尚无特异性上调SLC9A2表达的有效药物,亦缺乏针对STAT3通路激活的成熟靶向治疗。抑制STAT3上游信号成为可行策略,其中JAK为STAT3最关键的上游激酶。已有研究报道,JAK抑制剂Ruxolitinib可协同免疫治疗,提升霍奇金淋巴瘤及非小细胞肺癌的疗效[26,27]。鉴于Ruxolitinib亦能调控黑色素瘤细胞对抗PD-1的原发耐药[28],作者推测其可通过抑制STAT3通路模拟SLC9A2功能,从而逆转CRC免疫治疗耐药。剂量探索实验显示,每日30 mg/kg Ruxolitinib联合免疫检查点抑制剂(Ruxolitinib+ICI)抑瘤效果最佳,且未表现肝、肾或肺毒性(图8A-D)。瘤内Western blot证实,Ruxolitinib显著抑制STAT3磷酸化并下调VEGFA表达(图8E);ELISA亦显示VEGFA分泌减少(图8F)。此外,Ruxolitinib可刺激颗粒酶B及IFN-γ释放(图8G)。免疫组化结果显示,Ruxolitinib降低CD31⁺血管数量,增加CD8⁺ T细胞浸润,减少Ki67⁺增殖细胞,并提高TUNEL⁺凋亡细胞比例(图8H)。综上,Ruxolitinib联合抗PD-1方案在CRC治疗中具有潜在的临床转化价值。
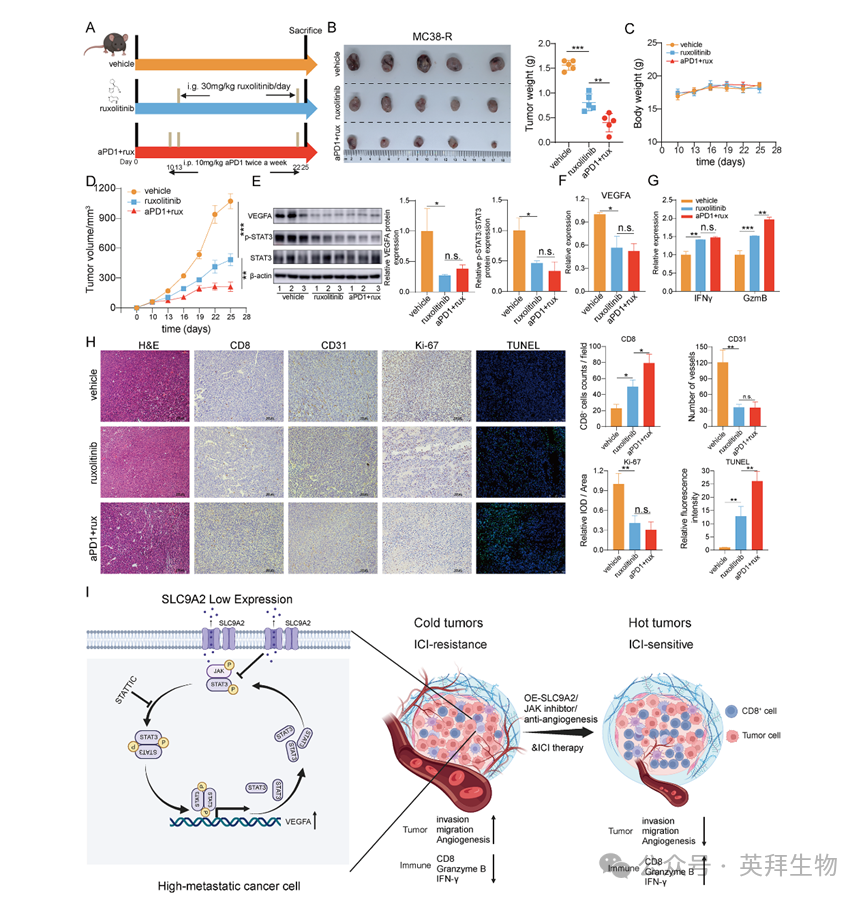
图8:Ruxolitinib增强抗PD-1免疫治疗结直肠癌的疗效
参考文献:
Zhang, Z., Liu, S., Xu, T., Chen, N., Liu, C., Yang, H., Shi, Y., Li, Z., Feng, X., Yao, Y., Duan, X., Xu, G., Zhang, C., Wang, Z., Li, J., & Shen, L. (2025). Tumor suppressor SLC9A2 inhibits colorectal cancer metastasis and reverses immunotherapy resistance by suppressing angiogenesis. Journal of experimental & clinical cancer research : CR, 44(1), 172.13046-025-03422-7