






CD160通过调控结直肠癌中CD8+ T细胞耗竭决定抗PD-1免疫治疗耐药性
结肠发生肿瘤的倾向性高于回肠,但免疫微环境差异在此现象中的作用尚不明确。本研究通过比较结直肠癌(CRC)患者和健康供体的配对回肠与结肠样本,发现回肠富集的CD160+CD8+ T细胞具有未被认知的特征:包括终末耗竭抗性和强大的克隆扩增能力。转移CD160+CD8+ T细胞可显著抑制微卫星高度不稳定(MSI-H)和炎症诱导的CRC模型中的肿瘤生长。Cd160基因敲除会加速肿瘤生长,而回输CD160+CD8+ T细胞可逆转该现象。值得注意的是,在MSI-H且抗PD-1耐药的CRC模型中,CD160+CD8+ T细胞通过增加肿瘤浸润性祖细胞耗竭T细胞,显著提升抗PD-1疗效并克服耐药性,几乎完全清除肿瘤。机制上,我们发现CD160与PI3K(p85α)的相互作用通过AKT–NF-κB通路促进FcεR1γ和4-1BB表达,从而增强CD8+ T细胞毒性。本研究揭示CD160是CD8+ T细胞功能的关键调控因子,并提出转移CD160+CD8+ T细胞这一创新免疫治疗策略以克服抗PD-1耐药。本文于2025年9月发表于《Nature Cell Biology》,IF: 19.1。
技术路线
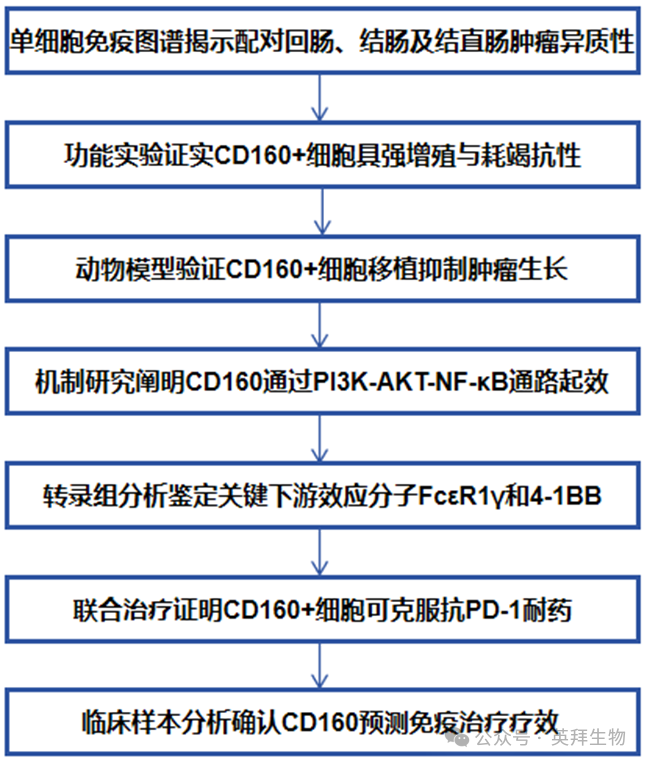
主要实验结果
1.单细胞免疫图谱揭示配对回肠、结肠及结直肠肿瘤的系统性差异
为构建免疫细胞图谱,我们获取了68例CRC患者和健康供体配对样本中CD45+免疫细胞的scRNA/BCR/TCR测序数据,包括肿瘤引流淋巴结(LNs)、外周血单核细胞(PBMCs)、回肠、结肠和肿瘤组织(图1a)。经过严格质量过滤后,保留465,920个高质量细胞。对多组学数据进行批次校正和无监督聚类后,我们鉴定出7个主要谱系,包括4个淋巴系亚型(T细胞、B细胞、自然杀伤(NK)细胞和浆细胞(PCs))及3个髓系亚型(肥大细胞、浆样树突状细胞(pDCs)和其他髓系细胞)(图1b)。配对的BCR和TCR分析结果与细胞鉴定高度一致。对T细胞的进一步亚群聚类发现了上皮内淋巴细胞(IEL)-T细胞的存在,其在回肠中含量最丰富。
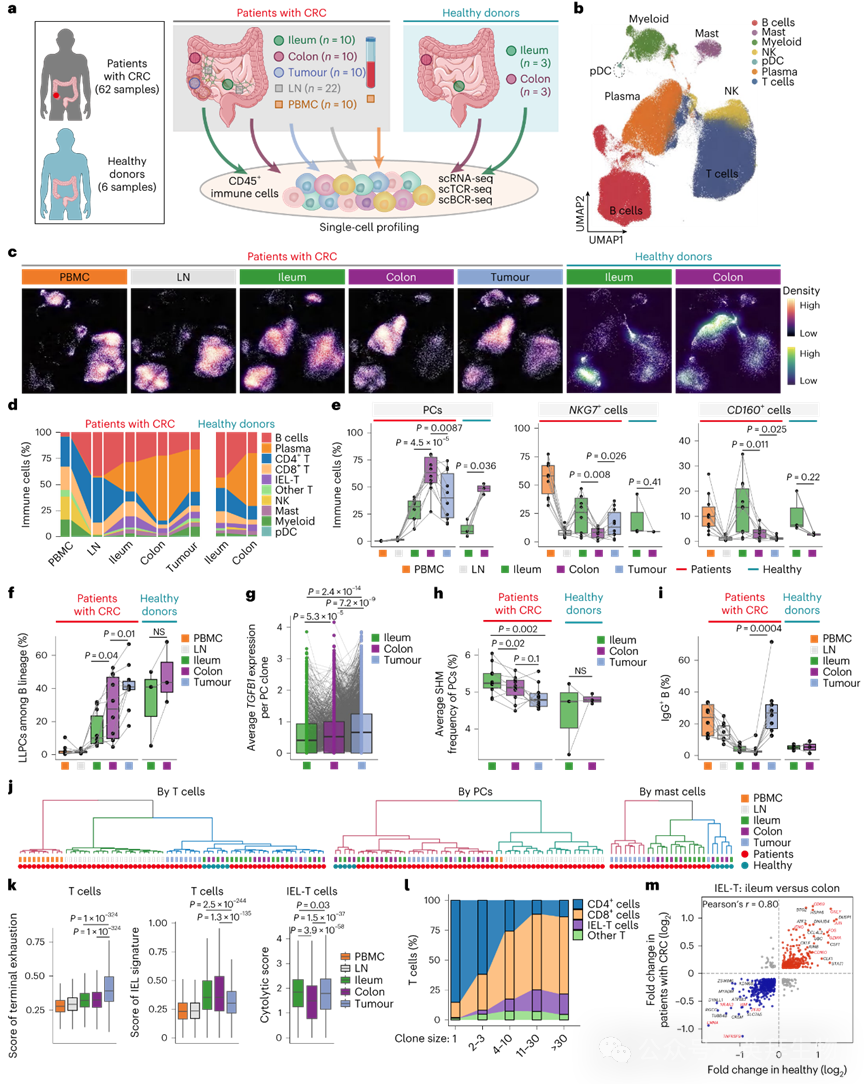
图1 配对回肠、结肠及结直肠肿瘤的单细胞免疫图谱
各组织间主要细胞亚群的组成和分布存在显著差异。PBMCs中富含T细胞和NK细胞,肿瘤引流淋巴结和回肠中富含B细胞,而肿瘤样本中髓系细胞富集,表明不同组织存在异质性免疫景观(图1c,d)。细胞毒性及T细胞活化标志物NKG7在回肠和肿瘤组织中的表达高于结肠组织(图1e)。与IEL-T细胞富集一致,CD160+细胞比例在回肠中最高,在结肠和肿瘤中急剧下降(图1e)。我们还鉴定出TGFB1high长寿命浆细胞(LLPCs)亚群,其在肿瘤和结肠组织中高度富集(图1f),这与浆细胞通过分泌转化生长因子(TGF)-β发挥调控作用一致。对跨组织3,188个PC克隆的BCR克隆匹配分析显示,同一克隆的PC在肿瘤中TGFB1表达最高,回肠中最低(图1g)。回肠来源的PCs具有最高的BCR体细胞超突变(SHM)频率,而肿瘤来源的PCs频率最低(图1h)。此外,与回肠或结肠相比,肿瘤来源的B细胞更倾向于IgG+表型(图1i),该趋势与细胞亚型组成无关,进一步表明B细胞在不同组织中可能呈现不同状态。髓系细胞中,单核细胞在肿瘤组织中积聚而肥大细胞减少。由于肥大细胞被激活时是抗肿瘤免疫的关键调控因子,我们构建了基因特征来量化肥大细胞的相对富集程度,其与CRC预后显著相关(风险比(HR)=0.78,95%置信区间0.66–0.93)。通过分别对每个细胞谱系进行无监督层次聚类,我们测量了跨组织的平均转录组相似性(图1j)。总体而言,PBMCs和肿瘤引流淋巴结的免疫细胞首先被区分开。所有细胞谱系中健康供体的组织样本聚集在一起,表明肿瘤对周围正常组织的免疫细胞状态具有显著影响。但肿瘤样本呈现高度异质性:部分与回肠相似度更高,另一部分则更接近邻近结肠组织。
使用已报道的基因特征集,我们发现肿瘤来源的T细胞表现出最高的终末耗竭特征,而回肠和结肠来源的T细胞显示最高的IEL特征(图1k)。通过scTCR-seq将T细胞克隆与细胞状态关联,我们观察到IEL-T细胞在扩增克隆中相较于其他T细胞显著富集(图1l)。此外,回肠来源的IEL-T细胞显示最高的细胞毒性活性(图1k)。为进一步探索该关联,我们比较了回肠与结肠组织中IEL-T细胞的转录组差异,发现健康供体与CRC患者间存在强相关性系统差异(图1m)。除CD160外,回肠中上调的基因包括T细胞活化相关基因(如IFNG、GZMA和GNLY)(图1m)。通过差异表达基因(DEGs)的平均表达对样本聚类,发现肿瘤来源的IEL-T细胞高度异质:部分样本的基因特征更接近结肠样IEL-T细胞,另一部分则更接近回肠样IEL-T细胞。
2.配对回肠、结肠及结直肠肿瘤中T细胞状态的异质性
为进一步探究肿瘤与回肠和结肠组织相关的异质性,我们比较了GTEx项目中回肠(n=187)和结肠(n=779)样本以及癌症基因组图谱(TCGA)项目中CRC样本(n=458)的转录组相似性。对免疫相关基因的无监督聚类将CRC样本分为两个簇:一个与结肠组织聚为一类(C1),另一个与回肠组织聚为一类(C2),其中C2的预后显著优于C1。C2与C1之间的差异表达基因(DEGs)在T细胞激活通路中显著富集,表明回肠与结肠间不同的T细胞状态在异质性肿瘤微环境(TME)中显现并影响预后。为在控制患者间异质性的前提下验证这一结果,我们获取了健康供体和CRC患者的配对回肠与结肠组织,证实了T细胞激活的富集现象。
为表征跨组织异质性T细胞状态,我们进行了进一步的聚类分析,揭示了6种CD4+ T细胞状态、8种CD8+ T细胞状态以及γδT细胞(图2a)。IEL-T细胞通过经典IEL标志物表达及肠道固有层标志物CD6缺失进行鉴定(图2a)。具有强IEL特征的γδT细胞通过TYROBP和TCRγδ基因的额外表达被识别,鉴于其与CD8+ T细胞的高度转录和功能相似性,将其归为CD8+ T细胞群(图2a)。本研究数据集中未检测到近期报道的TCRγδ+ CD8α+ IEL-T细胞群。总体而言,回肠中的CD4+和CD8+ T细胞显示T细胞活化相关基因上调,而肿瘤样本中T细胞功能障碍相关基因上调(图2b)。
在CD4+ T细胞中,调节性T细胞(Treg)和滤泡辅助T细胞(TFH)在引流淋巴结和肿瘤样本中更丰富,而初始T细胞(TNaive)、中央记忆T细胞(TCM)和效应记忆T细胞(TEM)在回肠和结肠中更丰富,且研究较少的CD4+细胞毒性T细胞(TCTL)在PBMCs和回肠中富集(扩展数据图3l)。与结肠和肿瘤相比,回肠表现出相对较高比例的TEM和TCTL细胞及较低比例的Treg细胞,尽管由于队列规模较小,并非所有差异均达到统计学显著性。在CD8+ T细胞中(图2a),记忆T细胞(TMem)在回肠中更丰富,而类似于先前描述的GZMK+效应T细胞的过渡效应T细胞(GZMK+ eff.)在肿瘤区域更丰富。为鉴定肿瘤反应性T细胞,我们采用基于已报道的新抗原反应性肿瘤浸润T细胞特征的肿瘤反应性特征谱,显示Treg、TFH和TCRαβ+ IEL-T细胞处于肿瘤反应状态。此外,健康供体回肠来源的Treg和TFH细胞具有最低的新抗原反应性评分,表明肿瘤发生影响周围回肠或结肠的CD4+ T细胞状态。
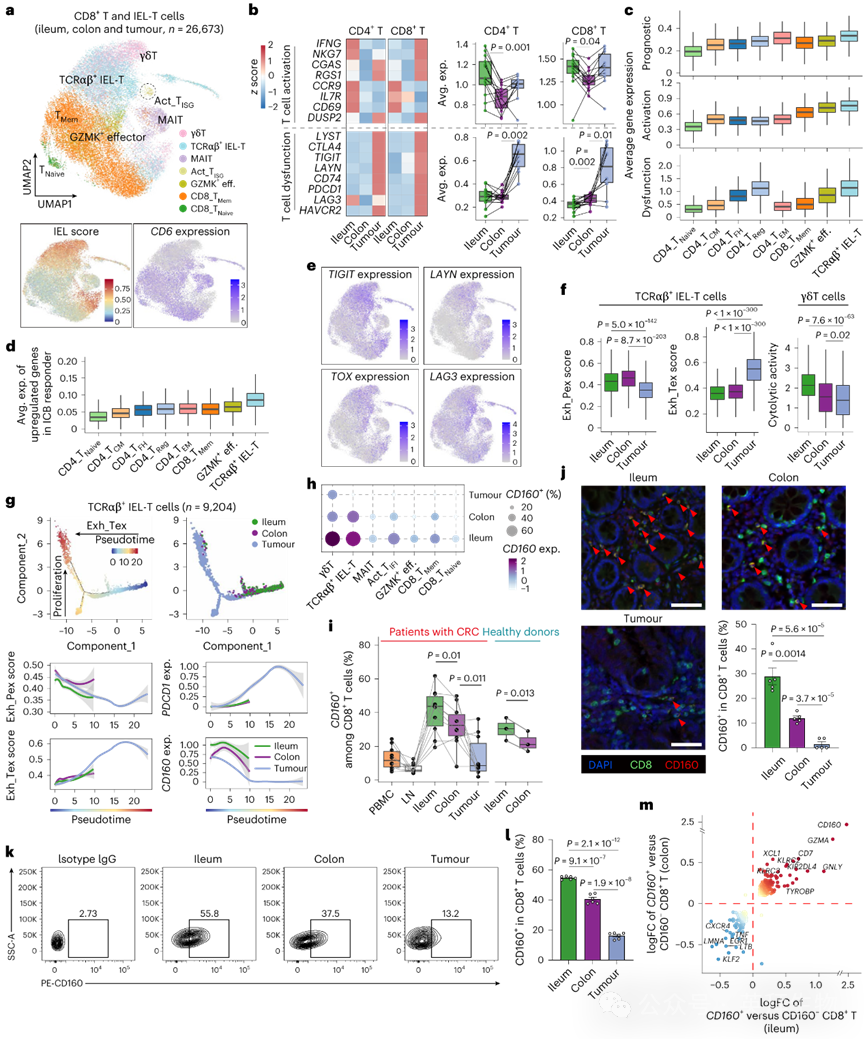
图2 健康供体与CRC患者回肠和结肠组织中T细胞亚群的免疫特征
我们随后测量了TCGA CRC样本中129个预测良好预后的基因在TME内主要T细胞状态中的平均表达,发现TCRαβ+ IEL-T细胞中表达最强(图2c)。TCRαβ+ IEL-T细胞还显示出最高的T细胞激活和功能障碍特征。一致的是,C2簇的CRC样本比C1簇表现出显著更高的IEL特征。我们从接受一线ICB治疗的CRC队列中获取了免疫检查点阻断(ICB)应答者中上调的基因,并观察到TCRαβ+ IEL-T细胞中的表达最强(图2d)。
我们随后聚焦于高表达T细胞耗竭标志物TIGIT、LAYN、TOX和LAG3的TCRαβ+ IEL-T细胞(图2e)。由于近期研究揭示了耗竭T细胞的异质性状态,包括对ICB应答的终末耗竭T细胞(TTex)和祖细胞耗竭T细胞(TPex),我们测量了单个细胞的祖细胞和终末耗竭CD8+ T细胞特征。通过跨组织比较,发现回肠和结肠来源的TCRαβ+ IEL-T细胞表现出显著更高的祖细胞耗竭(Exh_Pex)特征评分,而肿瘤来源的细胞表现出显著更高的终末耗竭(Exh_Tex)评分(图2f)。我们还比较了γδT细胞(既往研究报道其在结肠癌中细胞毒性降低并向终末耗竭状态转化),观察到γδT细胞在回肠中细胞毒性最高,在肿瘤中最低(图2f)。
3.CD160表达特征揭示CD8+ T细胞耗竭的状态转换
为研究耗竭T细胞状态转换,我们使用Monocle进行伪时间分析以推断TCRαβ+ IEL-T细胞的分化轨迹。分析显示轨迹始于回肠和结肠,随后分化为两个富集肿瘤来源T细胞的分支。轨迹成分1与Exh_Tex特征相关,成分2与细胞增殖信号相关(图2g)。沿伪时间轴,我们观察到Exh_Tex信号升高而Exh_Pex信号降低(图2g),与既往TPex细胞产生TTex细胞的报道一致。为进一步证实伪时间早期和晚期的TCRαβ+ IEL-T细胞分别对应更偏向祖细胞和终末耗竭的表型,我们检测了经典标志物的动态表达变化。一致地,与更终末耗竭状态相关的EOMES和TOX表达沿伪时间逐渐增加,而与祖细胞状态标志物IL7R和REL的表达水平沿伪时间逐渐降低。我们随后检测了其他非经典基因沿轨迹的表达动力学,发现CD160表达在所有三种组织中均呈现高度相关性变化,表明CD160表达与IEL-T细胞状态转换密切相关(图2g)。
CD160已被认为是IEL的标志物。然而,我们发现CD160在CD8+ T细胞亚群中广泛表达且表现出强烈的组织特异性(图2h)。在所有CD8+ T细胞亚型中,回肠的CD160+ T细胞比例最高而肿瘤中最低(图2i),与公共数据集一致。为进一步验证CD160的组织特异性表达,我们使用多重免疫荧光和流式细胞术在独立CRC样本中定量CD160+ CD8+ T细胞,观察到回肠中比例最高而肿瘤中最低(图2j-l)。类似地,在C57BL/6小鼠中,小肠(十二指肠、空肠和回肠)的CD160+ CD8+ T细胞显著高于结肠。我们在AOM/DSS诱导的CRC小鼠中进一步证实这一发现,发现回肠CD8+ T细胞中CD160表达最高而肿瘤CD8+ T细胞中最低。这些结果与我们的scRNA-seq数据一致,表明CD160表达可能受组织局部环境动态调控。回肠和结肠中CD160+与CD160− CD8+ T细胞的比较显示,涉及T细胞毒性的基因均一致性上调(图2m),与已报道的CD160+ T细胞溶细胞效应相符。为探究CD160是否表征T细胞耗竭的状态转换,我们比较了单个效应CD8+ T细胞(包括TCRαβ+ IEL-T和GZMK+效应细胞)的祖细胞与终末耗竭特征。CD160表达与Exh_Pex特征呈正相关,与Exh_Tex特征呈负相关(图3a)。这些数据共同支持CD160表达与CD8+ T细胞耗竭状态转换的关联性。
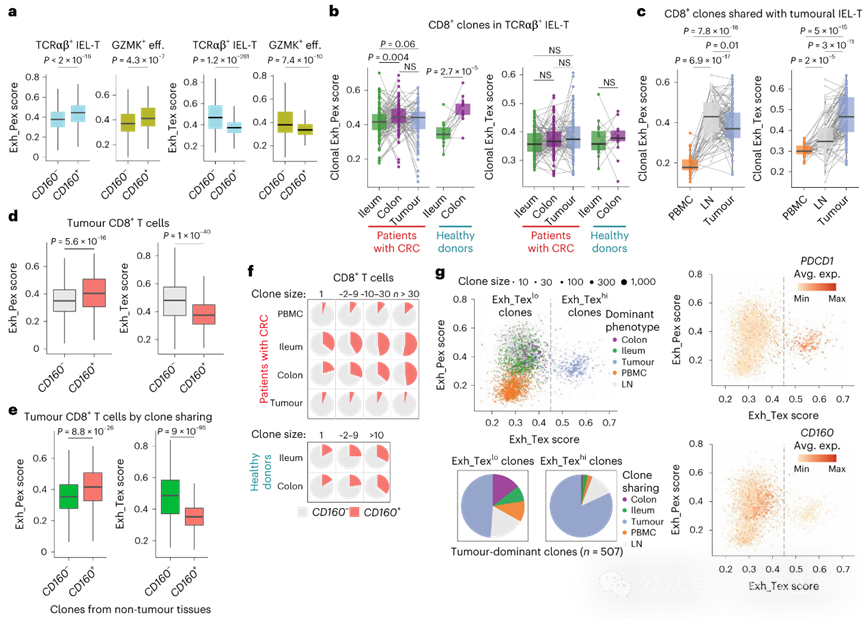
图3 CD8+ T细胞克隆状态转换的谱系追踪
4.肿瘤耗竭CD8+ T细胞与CD160− T祖细胞存在克隆关联
为评估IEL-T细胞克隆状态是否因组织而异,我们通过TCRαβ+ IEL-T细胞的克隆匹配分析评估其耗竭特征。值得注意的是,在回肠、结肠和肿瘤组织间匹配的大多数TCRαβ+ IEL-T克隆均含有CD160+ T细胞。虽然从回肠到结肠及肿瘤的匹配克隆中Exh_Pex特征平均值略有升高,但Exh_Tex特征未显著增加(图3b)。我们还比较了与肿瘤TCRαβ+ IEL-T细胞克隆匹配的PBMCs和淋巴结T细胞,发现从PBMCs和淋巴结到肿瘤区域的克隆中Exh_Tex特征显著增加(图3c)。此外,我们观察到淋巴结中克隆关联T细胞的Exh_Pex特征最高(图3c),这与近期报道的肿瘤中TTex细胞源于淋巴结祖细胞/干细胞样耗竭T细胞的观点一致。我们还根据克隆匹配T细胞的组织来源对肿瘤TCRαβ+ IEL-T细胞进行分组。与回肠或结肠共享克隆的细胞相比,肿瘤特异性或与PBMCs及淋巴结T细胞克隆关联的肿瘤IEL-T细胞具有显著更高的Exh_Tex特征和更低的Exh_Pex特征。这些数据共同表明,相对于淋巴结和PBMCs来源的T细胞,回肠和结肠来源的CD160+ CD8+ T细胞在肿瘤中不易发生终末耗竭。
为进一步探索CD160与T细胞耗竭的关联,我们根据CD160表达情况比较了肿瘤CD8+ T细胞的耗竭评分,发现CD160− T细胞比CD160+ T细胞具有更高的Exh_Tex特征和更低的Exh_Pex特征(图3d)。该模式在一个独立的CRC scRNA-seq数据集中得到进一步证实。接下来,我们使用非肿瘤组织中的所有CD8+ T细胞定义CD160+和CD160−克隆,并比较与这些细胞克隆关联的肿瘤CD8+ T细胞的耗竭特征。与非肿瘤CD160−克隆关联的肿瘤CD8+ T细胞相比,与非肿瘤CD160+克隆关联的细胞具有显著更高的Exh_Pex特征和更低的Exh_Tex特征(图3e和扩展数据图4o)。由于细胞毒性CD8+ T细胞克隆优先扩增,我们随后通过按克隆大小分类T细胞来检验CD160是否表征克隆扩增。我们注意到在正常组织(而非肿瘤)中,扩增克隆内CD160+ T细胞比例逐渐增加(图3f),表明CD160+ T细胞在不同组织中具有 distinct 的克隆扩增偏好。
对所有CD8+ T细胞单个克隆进行表征后,我们发现在肿瘤T细胞主导的最大克隆中Exh_Tex特征较高,而在PBMCs T细胞主导的克隆中Exh_Pex特征较低(图3g)。总体而言,具有高Exh_Pex特征的克隆明显由回肠/结肠主导克隆和淋巴结主导克隆区分。我们根据克隆分布的自然截断值将肿瘤CD8+ T细胞主导的所有克隆分为Exh_Texhi和Exh_Texlo两组,并分析其与其他组织的谱系关系(图3g)。发现大多数Exh_Texlo克隆与其他组织存在谱系关联,而Exh_Texhi克隆主要局限于肿瘤或与淋巴结CD8+ T细胞克隆关联。将单个肿瘤CD8+ T细胞分为Exh_Texhi和Exh_Texlo两组后,我们观察到肿瘤Exh_Texhi细胞与PBMCs来源的CD8+ T细胞存在显著谱系关联,表明这些细胞未包含在肿瘤CD8+ T细胞主导的克隆中。另一方面,极少肿瘤Exh_Texhi细胞与回肠和结肠的CD8+ T细胞存在克隆关联。这些结果表明TTex细胞要么是肿瘤特异性的,要么与淋巴结来源的CD160− CD8+ T细胞克隆关联。进一步的转录组比较表明,健康供体和CRC患者不同组织来源的CD160⁺CD8⁺ T细胞保持转录稳定性且不易发生终末耗竭,而CD160− CD8⁺ T细胞上调Exh_Tex特征。在MC38荷瘤小鼠中,脾脏、回肠、结肠和肿瘤组织中的CD160⁺CD8⁺ T细胞比CD160⁻细胞表现出显著更高比例的祖细胞耗竭细胞(TCF1+ PD-1+)。这些发现共同证明,不同组织来源的CD160⁺CD8⁺ T细胞具有高度相似的转录组特征和祖细胞耗竭表型。
5.CD160界定具有强细胞毒性和耗竭抗性的CD8⁺ T细胞亚群
为评估CD160+ CD8+ T细胞功能,我们比较了CD160+与CD160− CD8+ T细胞的细胞毒性颗粒表达。抗CD3/CD28刺激后,CD160+ CD8+ T细胞的GzmB+比例高于CD160−细胞(图4a,b),表明其细胞毒性更强。为进一步研究CD160+ CD8+ T细胞的耗竭抗性,我们通过持续抗CD3刺激建立体外慢性刺激模型(该模型诱导耗竭标志物PD-1和TIM3上调)(图4c),发现CD160+ CD8+ T细胞比CD160−细胞表现出更少的终末耗竭(TIM3+ PD-1+)表型(图4d,e),证实其增强的终末耗竭抗性。接下来,我们从MC38-GFP荷瘤小鼠腋窝淋巴结中分离CD160+和CD160− CD8+ T细胞并分别扩增(图4f)。与自体肿瘤细胞共培养后发现,CD160+ CD8+ T细胞组的肿瘤细胞死亡显著更高(通过LIVE/DEAD指示染料检测)(图4g,h)。
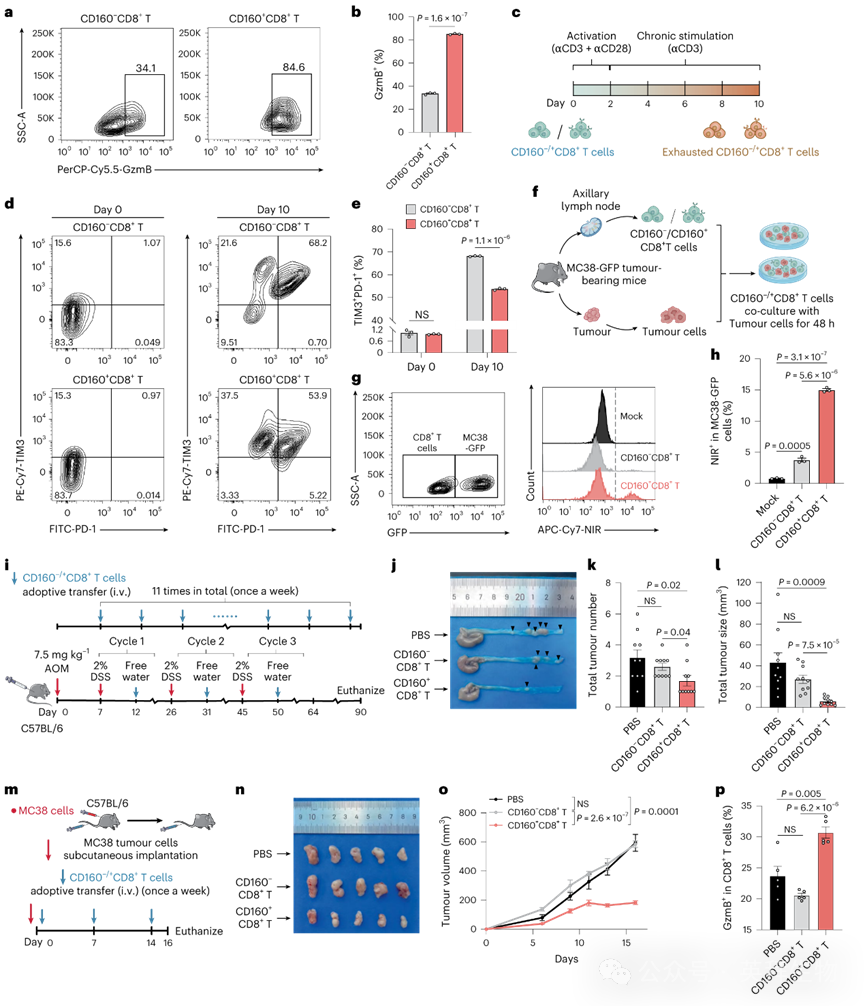
图4 CD160+ CD8+ T细胞具有强细胞毒性和耗竭抗性,过继转移可抑制CRC进展
为评估体内肿瘤浸润情况,我们将CD160+ CD8+ T细胞(来自GFP小鼠)和CD160− CD8+ T细胞(来自CD45.1小鼠)共同过继转移至CD45.2 MC38荷瘤小鼠。转移36小时后,观察到肿瘤内CD160+ CD8+ T细胞(GFP+ CD45.1−)的浸润显著高于CD160− CD8+ T细胞(GFP− CD45.1+),表明CD160+ CD8+ T细胞具有增强的肿瘤浸润能力。为评估肿瘤特异性激活,将分选的CD45.1小鼠来源CD160+ CD8+ T细胞转移至MC38-OVA荷瘤小鼠。在肿瘤浸润的CD45.1+细胞中,OVA四聚体+细胞比例随时间依赖性增加,表明CD160+ CD8+ T细胞在TME中被激活并逐渐获得抗原特异性。
综上所述,CD160标记了具有细胞毒性和耗竭抗性的CD8+ T细胞,其兼具卓越的肿瘤浸润能力和在TME内逐步获得肿瘤抗原特异性的双重功能优势。这些发现表明CD160+ CD8+ T细胞在CRC肿瘤抑制中起重要作用。
6.过继转移CD160+ CD8+ T细胞通过增强肿瘤浸润CD8+ T细胞毒性抑制结直肠肿瘤发展
为探究CD160+ CD8+ T细胞的体内抗肿瘤效应,我们在AOM/DSS诱导的CRC小鼠模型中进行了过继转移实验(图4i)。与接受PBS的小鼠相比,接受CD160+ CD8+ T细胞的小鼠肿瘤数量和大小显著减少(图4j-l)。而接受CD160− CD8+ T细胞的小鼠未显示抗肿瘤效果。
接下来通过两种转移策略评估CD160+ CD8+ T细胞在不同肿瘤阶段的作用。首先在C57BL/6小鼠接种MC38细胞的同时注射CD160+ CD8+ T细胞以模拟其对早期肿瘤发展的影响(图4m),该组肿瘤体积显著小于CD160− CD8+ T细胞或PBS组(图4n,o)。此外,转移CD160+ CD8+ T细胞后,观察到肿瘤浸润CD160+CD8+和GzmB+CD8+ T细胞增加,表明其成功浸润TME并增强CD8⁺ T细胞毒性(图4p)。第二种策略在MC38接种7天、肿瘤形成后注射CD160+CD8+ T细胞,以评估其对晚期肿瘤发展的作用。同样,CD160+CD8+ T细胞在维持TME浸润和GzmB表达的同时抑制了肿瘤生长。值得注意的是,两组实验中CD160−CD8+ T细胞均未显示抗肿瘤效应(图4n)。
在低免疫原性CT26模型中,尽管CD160+CD8+ T细胞能浸润肿瘤,但未能抑制肿瘤生长,表明其抗肿瘤效应依赖于肿瘤免疫原性。
7.CD160缺失促进CD8+T细胞终末耗竭并削弱CD8+ T细胞介导的肿瘤控制
为探究CD160缺失对肿瘤控制的影响,我们构建了Cd160基因敲除(Cd160−/−)小鼠,并采用AOM/DSS和皮下MC38模型评估肿瘤进展(图5a,e)。两种模型中Cd160−/−小鼠均比野生型(WT)小鼠产生更多、更大的肿瘤。值得注意的是,过继转移同系CD160+CD8+T细胞可显著抑制Cd160−/−小鼠的肿瘤生长,而CD160−CD8+ T细胞无治疗效应(图5b–d,f,g)。此外,Cd160−/−小鼠肿瘤浸润GzmB+CD8+ T细胞减少,该现象可通过转移CD160+CD8+ T细胞得以挽救(图5h,i)。这些结果通过肿瘤切片的免疫组化(IHC)得到进一步证实(图5j,k)。
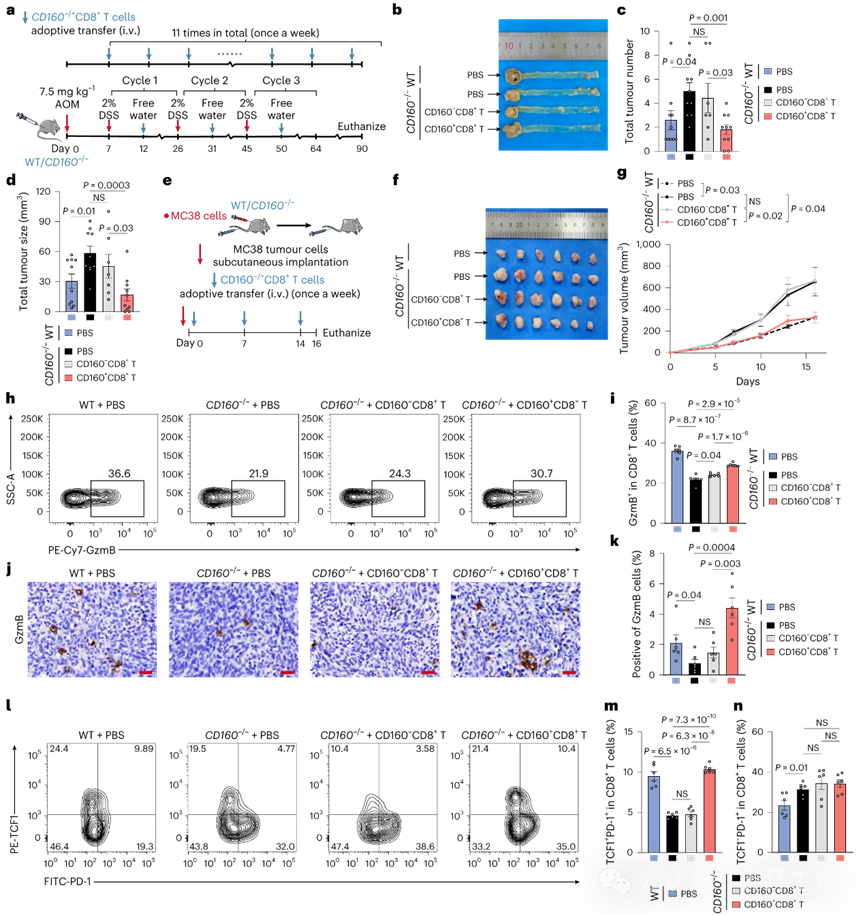
图5 CD160缺失加速肿瘤进展并促进肿瘤浸润CD8+ T细胞的终末耗竭
我们进一步分析了肿瘤浸润CD8+ T细胞的耗竭表型(祖细胞型:TCF1+ PD-1+;终末型:TCF1− PD-1+)。Cd160−/−小鼠表现为祖细胞耗竭CD8+ T细胞减少而终末耗竭细胞增多(图5l–n)。向Cd160−/−小鼠转移CD160+ CD8+ T细胞可大幅恢复TCF1+ PD-1+比例而不影响TCF1− PD-1+细胞比例,而转移相同数量的CD160− CD8+ T细胞无此效应(图5l–n)。这些结果共同表明CD160缺失会削弱CD8+ T细胞毒性、加速CRC进展并耗竭抗PD-1应答的祖细胞耗竭T细胞,支持CD160+ CD8+ T细胞在改善抗PD-1免疫治疗方面的 therapeutic potential。
8.过继转移CD160+ CD8+ T细胞赋予MSI-H CRC抗PD-1免疫治疗敏感性并克服耐药性
我们随后评估了转移CD160+ CD8+ T细胞是否能增强抗PD-1在MC38肿瘤中的疗效(图6a)。出乎意料的是,CD160+ CD8+ T细胞联合抗PD-1治疗可诱导近乎完全的肿瘤消退,效果优于任一单药治疗,而CD160− CD8+ T细胞未能增强抗PD-1疗效(图6b,c)。CD160+ CD8+ T细胞浸润肿瘤后增加了GzmB+ CD8+ T细胞比例,联合组尤为显著(图6d)。
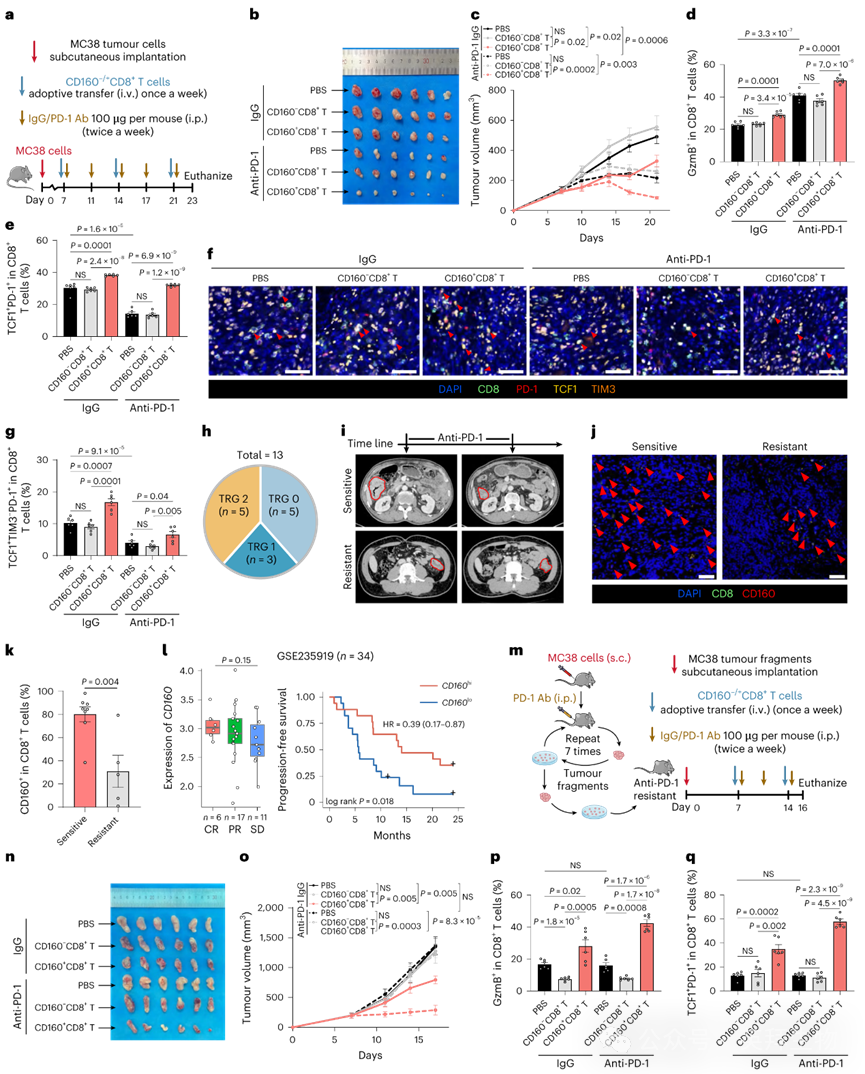
图6 肿瘤浸润CD160+ CD8+ T细胞赋予免疫治疗敏感性并克服MSI-H CRC的抗PD-1耐药
通过促使耗竭平衡向祖细胞耗竭T细胞倾斜以阻断抗PD-1治疗期间终末耗竭CD8+ T细胞的积聚对改善应答至关重要。我们发现CD160+ CD8+ T细胞单药或联合抗PD-1治疗均能显著提高肿瘤浸润CD8+ T细胞中TCF1+ PD-1+百分比,且不影响TCF1− PD-1+水平(图6e)。多重IHC进一步证实,单用CD160+ CD8+ T细胞或联合抗PD-1治疗的小鼠中祖细胞耗竭CD8+ T细胞增加(图6f,g)。在AOM/DSS诱导的CRC小鼠中,单用抗PD-1无明显疗效,但联合CD160+ CD8+ T细胞可显著降低肿瘤负荷。此外,单用CD160⁺CD8⁺ T细胞未能延长生存期,但与抗PD-1联用后可适度改善生存。
尽管抗PD-1免疫治疗对MSI-H型CRC患者具有良好疗效,但约50%此类患者存在抗PD-1耐药。为进一步确认CD160与抗PD-1应答的关系,我们收集了13例接受抗PD-1治疗的MSI-H型CRC患者。根据肿瘤退缩等级(TRG)定义应答:敏感组(TRG <2,n=8)与耐药组(TRG ≥2,n=5)(图6h)。CT扫描证实敏感组抗PD-1治疗后原发肿瘤持续缓解,而耐药组存在较大残留肿瘤(图6i)。确实,我们观察到敏感组肿瘤浸润CD8+ T细胞中CD160表达显著更高(图6j,k)。一致地,在独立接受一线ICB治疗的CRC队列中,CD160高表达预示改善的生存期(图6l),且该效应独立于CD8+ T细胞浸润。完全缓解者CD160表达更高,尽管因队列规模较小未达统计学显著性。
为进一步探究CD160+ CD8+ T细胞是否能克服MSI-H型CRC的抗PD-1耐药,我们通过多轮抗PD-1治疗直至抑瘤效应消失,建立了抗PD-1耐药的MC38肿瘤模型(图6m–o)。转移CD160+ CD8+ T细胞可恢复抗PD-1敏感性,导致肿瘤近乎完全消退(图6n,o)。该现象伴随肿瘤浸润GzmB+ CD8+ T细胞和祖细胞耗竭T细胞的增加(图6p,q)。而CD160− CD8+ T细胞无此效应。这些结果表明CD160+ CD8+ T细胞能赋予MSI-H型CRC免疫治疗敏感性并克服抗PD-1耐药。
9.CD160–PI3K相互作用通过促进FcεR1γ和4-1BB表达增强CD8+ T细胞毒性
为探究CD160在CD8+ T细胞中的功能,我们比较了从小鼠脾脏分选的CD160+与CD160− CD8+ T细胞的转录组。CD160+ CD8+ T细胞显示Fcer1g显著上调(图7a),该基因以转导免疫受体激活信号而闻名。基因本体(GO)富集分析显示T细胞激活、NF-κB调控和T细胞介导的细胞毒性通路均上调(图7b)。在T细胞激活通路相关基因中,参与CD8+ T细胞存活和记忆形成的CD160下游效应分子Tnfrsf9在CD160+ CD8+ T细胞中高度上调(图7c)。
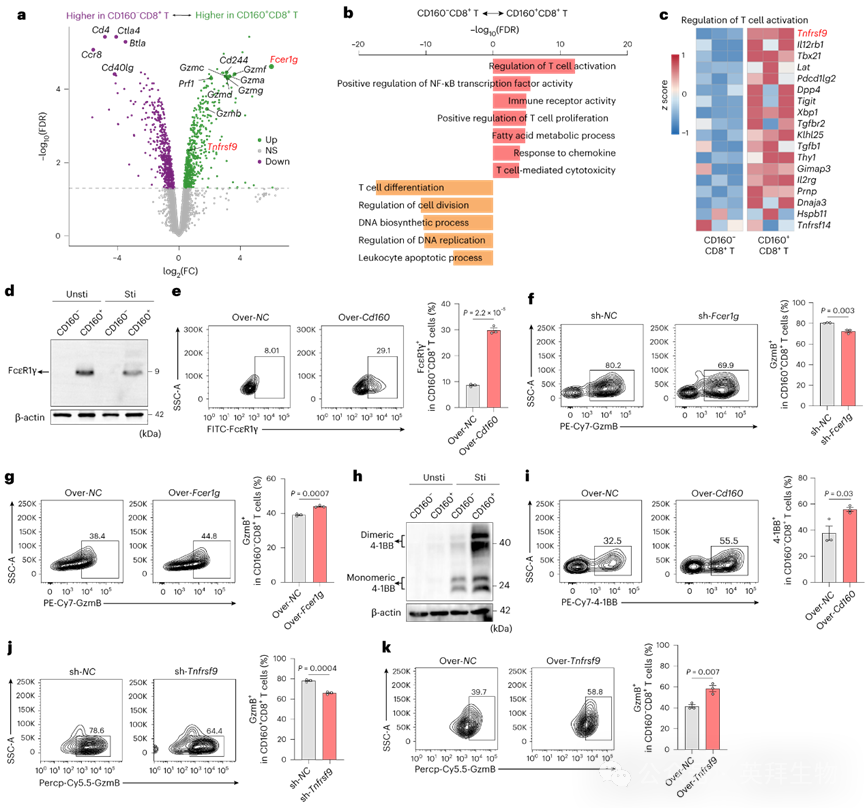
图7 CD160通过上调FcεR1γ和4-1BB表达增强CD8⁺ T细胞毒性
在mRNA/蛋白水平上,CD160+ CD8+ T细胞中Fcer1g(编码FcεR1γ)表达显著更高(图7d)。在CD160− CD8+ T细胞中过表达Cd160可诱导FcεR1γ表达,而敲低Fcer1g会降低CD160+ CD8+ T细胞的GzmB表达,削弱其细胞毒性。相反,过表达Fcer1g可增强CD160− CD8+ T细胞毒性(图7e–g)。类似地,CD160+ CD8+ T细胞中Tnfrsf9(编码4-1BB)表达升高,且Cd160过表达可诱导4-1BB表达(图7h,i)。沉默Tnfrsf9会降低CD160+ CD8+ T细胞的GzmB水平,而过表达Tnfrsf9可增强CD160− CD8+ T细胞毒性(图7j,k)。同时,CRC患者和MC38荷瘤小鼠的肿瘤浸润CD160+ CD8+ T细胞仍维持显著高于CD160− CD8+ T细胞的FcεR1γ和4-1BB表达。这些结果表明FcεR1γ和4-1BB是CD160增强CD160+ CD8+ T细胞毒性的关键下游效应分子。
我们进一步探究了CD160如何调控CD8+ T细胞中Fcer1g和Tnfrsf9表达。NF-κB是Fcer1g和Tnfrsf9的转录因子,GO分析显示NF-κB信号在CD160+ CD8+ T细胞中富集。与CD160− CD8+ T细胞相比,CD160+ CD8+ T细胞上调了经典NF-κB通路(p65)而非非经典通路(p52)。免疫荧光及核质蛋白印迹显示NF-κB p65在CD160+ CD8+ T细胞中发生显著核转位,而在CD160− CD8+ T细胞中未能入核(图8a–c)。用JSH-23阻断NF-κB核转位可降低Fcer1g和Tnfrsf9表达,表明CD160通过NF-κB促进其转录(图8d)。
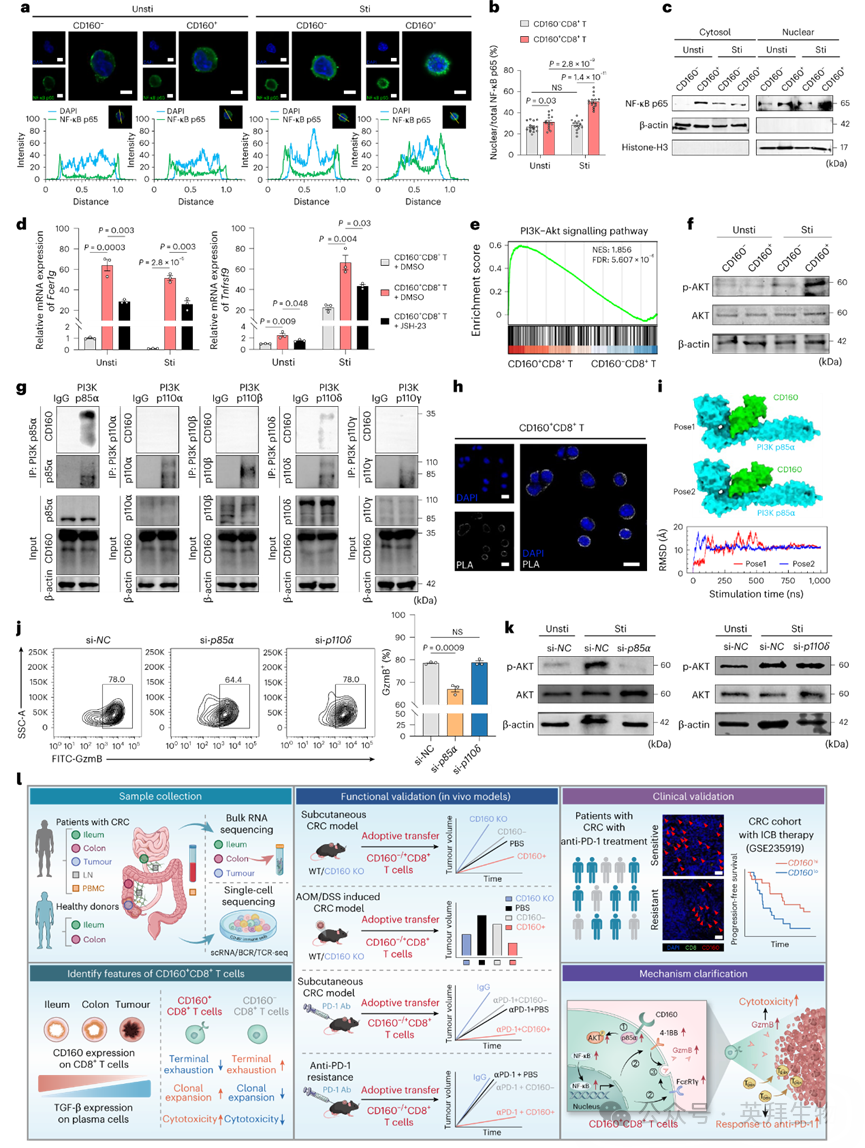
图8 CD160与PI3K p85α相互作用促进NF-κB核转位
通过基因集富集分析(GSEA)发现,对T细胞激活、增殖、毒性及NF-κB核转位至关重要的PI3K-AKT通路在CD160+ CD8+ T细胞中上调(图8e),且p-AKT水平升高(图8f)。I类PI3K是由催化亚基(p110)和调节亚基(p85)组成的异二聚体。为确定CD160是否通过与PI3K亚基直接相互作用激活PI3K–AKT通路,我们在CD160⁺CD8⁺ T细胞中进行了内源性免疫共沉淀(co-IP)。值得注意的是,在相同曝光条件下,CD160与p85α强结合,与p110δ弱结合,未检测到与p110α、p110β或p110γ的相互作用(图8g)。该相互作用特异性在共转染HA标签CD160与Flag标签PI3K p85α或p110δ的HEK293T细胞中得到证实,其中HA-CD160优先结合p85α。邻近连接检测(PLA)进一步揭示CD160⁺CD8⁺ T细胞中存在CD160–p85α直接相互作用,而CD160−细胞中无此现象(图8h)。分子动力学模拟与对接分析显示,CD160–p85α复合物比其他PI3K亚基复合物具有更高结合稳定性(图8i)。功能上,敲低CD160+ CD8+ T细胞中的p85α会消除CD160介导的GzmB表达和AKT磷酸化,而敲低p110δ无影响(图8j,k)。这些发现共同证明,CD160通过直接与p85α相互作用激活AKT–NF-κB通路,从而促进FcεR1γ和4-1BB表达,增强CD8+ T细胞毒性。
结论
总之,本研究提供了具有多区域和克隆分辨率的独特CRC患者单细胞资源。我们揭示了回肠高度富集的CD160+ CD8+ T细胞的组织特异性特征,证明了其在MSI-H型CRC中的治疗潜力——能够增强抗PD-1疗效并克服其耐药性,提示其在泛癌治疗中具有重要临床转化价值。
参考文献:
Zheng T, Ding C, Lai S, Gao Y, Lyu C, Liu C, Shi J, Li X, Li M, Meng H, Li M, Liang Y, Tai S, Cheng L, Zhang Y, Li L, Han P, Sun B, Liu T, Geng F, Hao D, Zhang X. CD160 dictates anti-PD-1 immunotherapy resistance by regulating CD8+ T cell exhaustion in colorectal cancer. Nat Cell Biol. 2025 Sep 9. doi: 10.1038/s41556-025-01753-3. Epub ahead of print. PMID: 40925954.