






MT-125通过抑制非肌肉肌球蛋白IIA与IIB延长胶质母细胞瘤生存期
胶质母细胞瘤(GBM)是最致命的原发性脑肿瘤。本研究报道了一种非肌肉肌球蛋白II的小分子抑制剂MT-125。该化合物具有高脑渗透性和优异的安全性特征,能阻断GBM侵袭和胞质分裂,延长小鼠GBM模型的生存期。通过损害线粒体分裂,MT-125可增加氧化应激及随之产生的DNA损伤,并与放疗产生协同效应。在氧化应激驱动机制下,MT-125还会诱导肿瘤细胞对PDGFR信号传导产生癌基因成瘾性,并与FDA批准的PDGFR及mTOR抑制剂在体外产生协同作用。与此一致的是,我们发现将MT-125与PDGFR抑制剂舒尼替尼或磷脂酰肌醇3-激酶(PI3K)/mTOR双重抑制剂帕沙利西联用,在原位GBM模型中的生存获益显著优于单药治疗。我们的研究结果表明,MT-125作为首创新药具有治疗GBM的强大临床潜力。本文于20205年8月发布于《Cell》, IF:42.5。
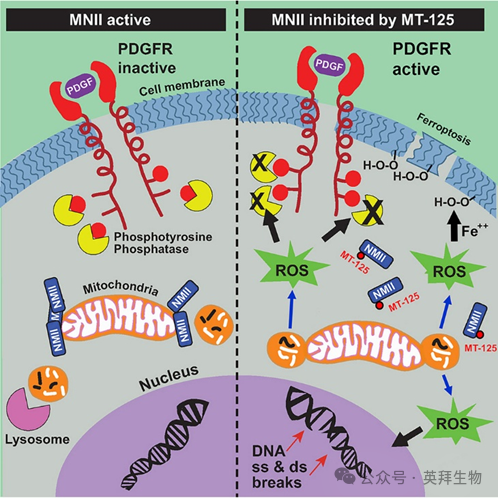
图形摘要
主要实验结果
1. MT-125是对NMIIA和NMIIB具有高选择性的布比斯塔汀类似物
通过评估基于布比斯塔汀衍生的NMII抑制剂化合物库,我们选择了MT-125(图1A)。该化合物对NMIIA(Ki,NMIIA = 2.7 ± 0.2 μM;图1B)和NMIIB(EC50 = 1.7 ± 0.1 μM;图1C)的抑制效力高度相似,在所有测试剂量下对正常细胞均无细胞毒性(图1C红色标注),且对心肌球蛋白II(CMII)的抑制活性极低(KI,CMII = 50 ± 10 μM;图1B)。我们前期工作预测这种对NMIIA/IIB的20-30倍选择性将转化为优异的体内耐受性。既往报道显示,骨骼肌肌球蛋白II特异性衍生物MT-134(对CMII抑制较弱,KI,CMII = 83 μM)在体内至少10 mg/kg的最高测试剂量下仍表现良好耐受性;另一种NMII抑制剂MT-228(KI,CMII = 17 μM)在剂量至少比布比斯塔汀(KI,CMII = 1.9 μM)高20倍时对心脏功能影响甚微。
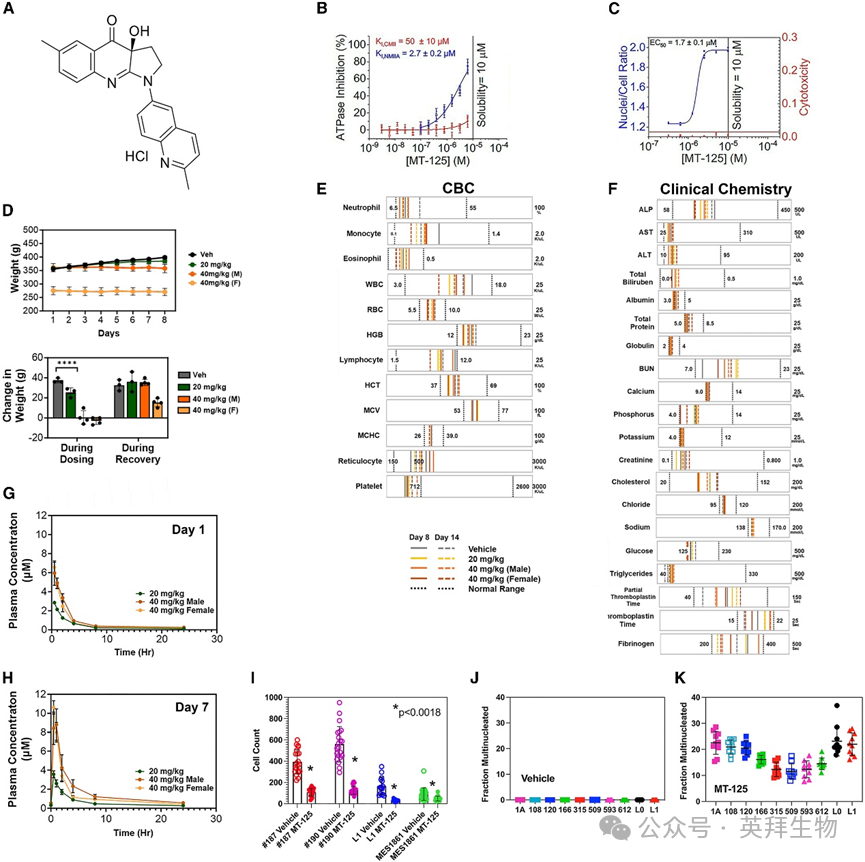
图1 MT-125具有NMIIA/IIB特异性、无毒性且可模拟NMII缺失表型
2.MT-125的药代动力学、代谢及安全性评价
我们分析了MT-125的类药物特性。微粒体稳定性研究预测其在体内具有相对较短的半衰期,这是平衡耐受性与有效性的理想特性。MT-125与肝微粒体蛋白孵育后的代谢物鉴定表明,其在非临床物种中的主要代谢途径与人类一致。此外,其细胞色素P450相互作用风险极低,且非P-糖蛋白底物,提示具有良好脑滞留性。在人醚相关基因-中国仓鼠卵巢细胞(hERG-CHO)电生理学检测中,MT-125的IC50估计值大于10 μM;在作为心脏毒性附加评估的离子通道活性组中,对hERG、Nav1.5、Kv4.3/KChiP2、KCNQ1/mink及Kir2.1的抑制率均低于25%。结合其对CMII的微弱抑制,这些结果表明MT-125不会影响心脏功能,且具有符合药物开发推进要求的选择性特征。
通过体内特性分析确定了MT-125的最佳给药途径、脑滞留程度、药代动力学特性及耐受性。静脉注射2 mg/kg时,MT-125的最大血药浓度(Cmax)为5.45 ± 0.18 μM,曲线下面积(AUClast)为4.46 ± 0.14 μM·h,血浆半衰期(T1/2)为10.3小时,且耐受性良好无临床不良反应。口服10 mg/kg同样耐受良好但生物利用度较低(Cmax = 0.72 ± 0.21 μM, AUClast = 1.44 ± 0.14 μM·h, T1/2 = 10.3 h)。因溶解度限制,更高口服剂量会形成悬浮液导致吸收不良。皮下注射(2、5和10 mg/kg)耐受性良好且系统暴露量优异,给药后短时间内脑部即达高浓度。例如5 mg/kg皮下注射后10分钟内(Tmax)脑内浓度达9.1 ± 1.0 μM,为同期血浆浓度的两倍(脑血浓度比B:P = 2.0)。此外,MT-125在注射后长时间内仍维持可检测的脑内浓度(脑AUClast = 5.4 ± 0.8 μM·h,T1/2 = 10.5 h)。我们比较了10 mg/kg皮下与腹腔注射30分钟后的血浆和脑药物浓度:腹腔给药的血浆浓度显著较低(腹腔 = 0.7 ± 0.3 μM,皮下 = 2.5 ± 0.2 μM)。但由于仅评估单时间点,这些数值不代表实际Cmax,且两种给药途径的药代动力学特征可能存在差异。但两种方式均显示优异脑渗透性(如10 mg/kg时脑浓度:皮下 = 3.7 ± 0.3 μM, B:P = 1.5;腹腔 = 1.0 ± 0.3 μM, B:P = 1.6)。
3.MT-125在体内表现出低毒性且具有良好治疗指数
小鼠每日皮下注射10 mg/kg MT-125连续两周耐受性良好,体重无显著变化(时间×处理交互作用:F(13, 91) = 1.5, p > 0.05),临床化学与血液学指标相较溶剂组均无异常(所有指标p > 0.05)。为确定最大耐受剂量(MTD),大鼠单次皮下注射60、70或90 mg/kg MT-125。给药1小时后采集血液进行毒代动力学分析,24小时后进行血液学及临床化学检测。所有剂量组均未观察到临床不良反应或体重变化(时间×处理交互作用:F(2, 6) = 1.1, p > 0.05)。给药1小时后血浆浓度表明MT-125已被吸收(60 mg/kg组:4.8 ± 0.6 μM;70 mg/kg组:3.6 ± 0.4 μM;90 mg/kg组:4.4 ± 0.3 μM)。但各剂量组浓度相近提示60 mg/kg可能是单次皮下注射的最大可吸收剂量。所有临床化学与血液学指标均在正常范围内。
我们进一步考察了溶剂与MT-125(20或40 mg/kg,皮下注射)每日重复给药7天的效果。溶剂组和20 mg/kg MT-125组均未观察到药物相关不良反应或对正常年龄相关性体重增长的影响(给药期间溶剂组vs 20 mg/kg组,p > 0.05;7天恢复期溶剂组vs 20 mg/kg组,p > 0.05;图1D)。雄性小鼠重复注射40 mg/kg后2-4小时出现轻微呼吸变化和流涎症状,均在30-60分钟内自行缓解。在此期间小鼠体重维持稳定并在恢复期增长(40 mg/kg雄性组:第1天vs第8天,p > 0.05;给药期间溶剂组vs 40 mg/kg组,p < 0.0001;7天恢复期溶剂组vs 40 mg/kg组,p > 0.05;图1D)。雌性小鼠未出现健康异常或体重变化(第1天vs第8天,p > 0.05;图1D)。治疗7天后及7天恢复期后,所有组的临床化学和血液学指标均正常(图1E、1F)。通过第1天(图1G)和第7天(图1H)0.5-24小时内6个时间点的毒代动力学评估(第7天加测给药前浓度),观察到预期的剂量依赖性血浆浓度升高,且无性别差异。40 mg/kg组雄性AUClast为20.4 ± 2.1 μM·h,雌性为17.0 ± 1.7 μM·h(p > 0.05)。给药7天后可见轻微蓄积现象,末次给药前血浆浓度为0.43 ± 0.05 μM(图1H)。
我们在良好实验室规范(GLP)条件下对雌雄大鼠进行了28天重复给药的毒理学研究,剂量为7.5、15和30 mg/kg(皮下注射)。在所有评估指标(包括临床观察、体重、摄食量、眼科检查、中枢神经系统功能观察组合测试(FOB)、临床病理学和组织病理学)中,MT-125均表现良好耐受性。所有动物在给药期间体重均增长,其中15和30 mg/kg组雄性大鼠体重增速稍缓,停药后恢复(双因素方差分析组间效应:雄性给药期间F(3,72)=3.1, p<0.05;第22天溶剂组vs 15 mg/kg组p<0.05,溶剂组vs 30 mg/kg组p<0.001;第28天溶剂组vs 15 mg/kg组p<0.001,溶剂组vs 30 mg/kg组p<0.0001;其余比较无显著性;雄性恢复期F(2,12)=2.5, p>0.05;雌性给药期间F(3,72)=0.5, p>0.05;雌性恢复期F(2,12)=2.9, p>0.05)。两性别均出现轻微超出正常范围的血糖和甘油三酯降低,其余指标均正常。毒代动力学测试确定30 mg/kg剂量对应Cmax=3.44 μM(给药后30分钟)和AUClast=15.40 μM·h。在28天给药期及后续28天恢复期内,所有剂量组均未记录到显著或持续的健康损害。由此确定未见不良反应剂量(NOAEL)为最高测试剂量30 mg/kg(皮下注射)。
4.MT-125可模拟NMII缺失对GBM侵袭与增殖的效应并延长小鼠GBM模型生存期
为验证MT-125抑制侵袭的效果是否与布比斯塔汀相当,我们检测了1个小鼠(MES1861)和3个人类(187、190和L1)GBM细胞系在溶剂(DMSO)或5 μM MT-125处理8小时后穿过3 μm Transwell小孔的体外侵袭能力(图1I)。MT-125在所有细胞系中均显著抑制Transwell侵袭(p < 0.0001)。NMII同时参与胞质分裂过程,其抑制会导致多核化和非整倍体形成。与此一致,5 μM MT-125处理48小时后,10个人类GBM细胞系中12%-25%的细胞出现多核化现象(图1J、1K)(所有MT-125处理组vs溶剂组p < 0.0001,双尾t检验)。
为探究MT-125对细胞核大小和数量的影响能否作为体内药物-靶点结合的生物标志物,我们采用基因工程小鼠模型(GEMM)——通过向携带条件性敲除肿瘤抑制基因(如Trp53或Pten)的小鼠原位注射编码PDGFbb-HA融合蛋白和cre重组酶的反转录病毒载体构建。该模型可100%形成 proneural 型、异柠檬酸脱氢酶(IDH)野生型、O6-甲基鸟嘌呤-DNA甲基转移酶(MGMT)非甲基化的GBM,且具有完全致死性。我们对Trp53(‒/‒)肿瘤小鼠每日腹腔注射溶剂或10 mg/kg MT-125连续14天,取脑组织切片进行H&E染色(图2A、2B)。MT-125诱导了多核化现象(图2B白色箭头)并使平均核横截面积增加53%(图2C, p < 0.0001)。我们另用溶剂或MT-125处理Trp53、Pten单敲或双敲除的小鼠GBM细胞系及7个人类GBM细胞系120小时,采用ATP检测法(CellTiter Glo,Promega)评估细胞毒性(图2D)。其中三个人类系(L0、L1和GBM1A)具有肿瘤起始细胞特征(图2E)。各细胞系的细胞毒效应EC50值为1-5 μM。结合药代动力学研究中的脑内浓度数据,我们预测2-10 mg/kg剂量在体内应具有疗效。
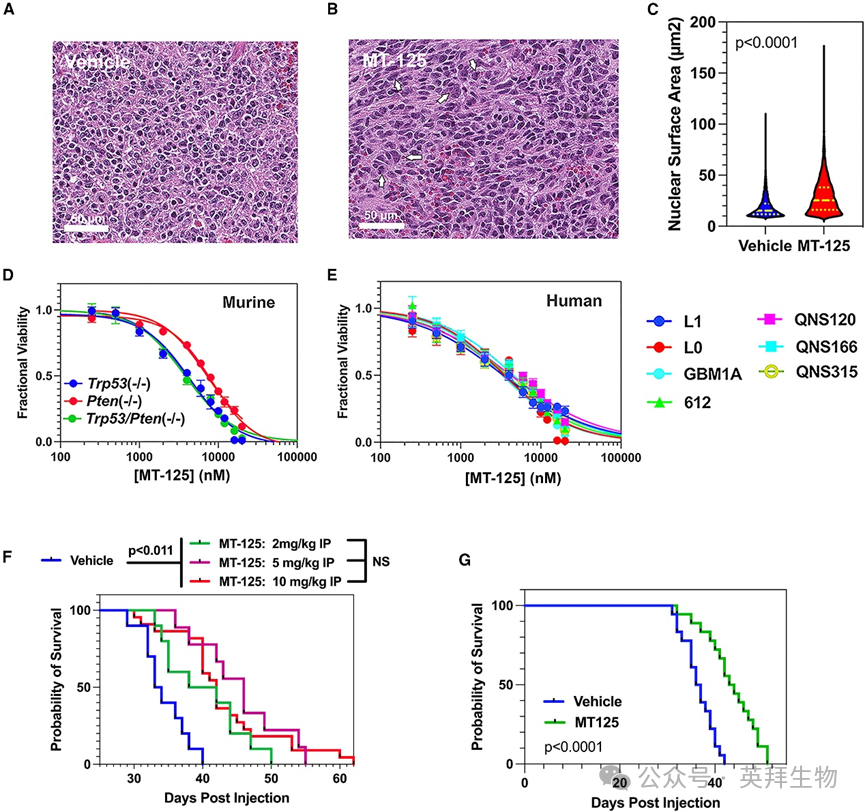
图2 MT-125对GBM具有治疗活性
为评估MT-125的治疗潜力,我们在反转录病毒原位注射7天后开始,每日对Trp53(‒/‒) GEMM模型进行腹腔注射溶剂或2、5、10 mg/kg MT-125(图2F),持续治疗至发病。获得的 Kaplan-Meier生存曲线表明,三种剂量MT-125单药治疗均较溶剂组延长中位生存期(三种剂量vs溶剂组p < 0.011,对数秩检验)。尽管存在剂量递增延长生存期的趋势,但未达到统计学显著性。我们另通过皮下注射2 mg/kg MT-125验证其在该模型中的疗效,同样显著延长生存期(图2G;p < 0.0001,对数秩检验)。因此将2 mg/kg设定为最低有效剂量(MED)。雌雄小鼠皮下注射2 mg/kg的药代动力学测试显示,该剂量下的生存获益仅需0.98 ± 0.2 μM·h的血浆AUClast浓度支持。给药30分钟后脑内MT-125浓度为0.4 ± 0.04 μM,且无性别差异。结合毒理学数据显示每日给药28天的NOAEL >30 mg/kg(图1D–1F),皮下或腹腔注射低至2 mg/kg剂量即可产生疗效,表明MT-125具有优于15倍的长期给药治疗指数。
5.MT-125诱导氧化还原介导的细胞毒性及放射增敏效应
约10 μM剂量MT-125(图2D、2E)在多种鼠源及人源GBM细胞系中产生>90%细胞毒性,但对正常细胞无影响(图1C)。鉴于DNA断裂是多种细胞死亡形式的特征,我们检测了MT-125对裂解聚 ADP 核糖聚合酶(PARP)与磷酸化H2AX(γH2AX)水平的影响。在MT-125处理的Trp53(‒/‒)细胞中(图3A),两者均升高且可被抗氧化剂N-乙酰-L-半胱氨酸(NAC)逆转。此外,敲除NMIIA或NMIIB足以使γH2AX增加6倍以上(图3B),表明该效应非脱靶作用。MT-125还在5种低传代人类GBM细胞系中增加γH2AX(图3C)。共济失调毛细血管扩张突变(ATM)蛋白响应DNA损伤磷酸化H2AX,ATM抑制剂AZD1390在鼠源Trp53(‒/‒) GBM细胞系中阻断了MT-125诱导的γH2AX升高(图3D)。这些变化与裂解 caspase 3的表达同步出现(图3E),提示MT-125诱导细胞凋亡。
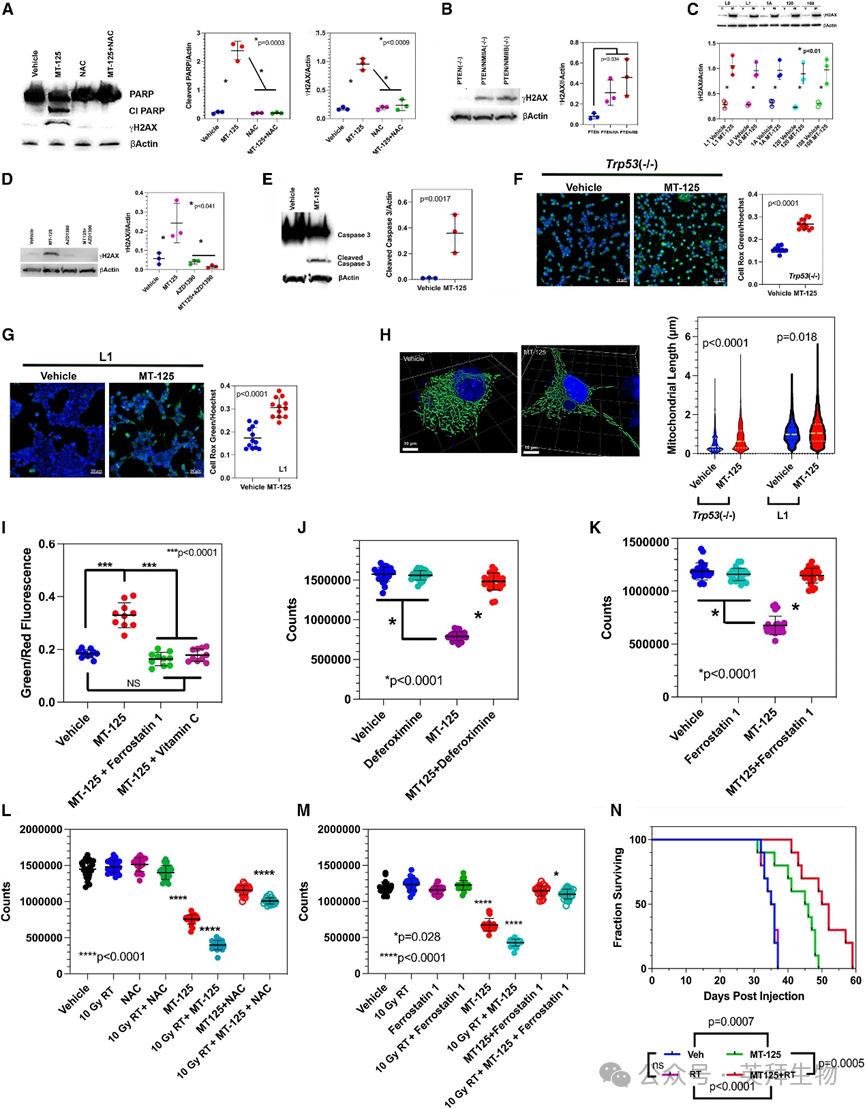
图3 MT-125诱导氧化还原介导的细胞毒性及放射敏感性
NMII的线粒体分裂调控功能可解释上述发现——该质量控制机制可清除受损线粒体膜,防止活性氧(ROS)泄漏至胞质与细胞核。为验证MT-125是否增加胞质ROS,我们在ROS敏感荧光探针CellRox Green存在下处理鼠源Trp53(‒/‒)和人源L1 GBM细胞,并用Hoechst复染。MT-125使鼠源(图3F)和人源细胞系(图3G)的标准化CellRox Green信号增强67%-77%。若该ROS升高源于MT-125抑制线粒体分裂,则其应同时延长线粒体长度。我们使用MitoTracker Green染色鼠源和人源GBM细胞系,每组测量8个细胞的线粒体长度。MT-125在两类细胞系中均显著增加线粒体长度(图3H)。ROS还可通过铁死亡过程过氧化膜脂损害细胞膜完整性,该过程需ROS和氧化还原活性铁共同参与。为验证MT-125是否诱导铁死亡,我们用定位细胞膜的荧光探针BodipyC11(脂质过氧化时荧光发射从红移向绿光)处理鼠源GBM细胞。与溶剂相比,EC50浓度(4 μM)MT-125处理48小时使绿/红荧光比率增加近一倍(图3I),该效应可被铁死亡抑制剂ferrostatin 1或ROS清除剂维生素C逆转。为探究铁死亡抑制剂是否阻断MT-125细胞毒性,我们用溶剂、两种铁死亡抑制剂(螯合氧化还原铁的deferoxamine和ferrostatin 1)、4 μM MT-125或4 μM MT-125+铁死亡抑制剂处理鼠源Trp53(‒/‒) GBM细胞120小时,并通过CellTiter Glo检测细胞活力。结果如图3J(deferoxamine)和图3K(ferrostatin 1)所示,两种抑制剂均几乎完全逆转MT-125的细胞毒性。
放射治疗作为GBM标准疗法,通过多种机制杀伤肿瘤细胞,其中部分机制依赖ROS生成(包括铁死亡),提示MT-125可能与放疗协同。我们在鼠源Trp53(‒/‒) GBM细胞系中开展体外验证(图3L、3M)。10 Gy照射、NAC(图3L)或ferrostatin 1(图3M)处理对细胞活力无显著影响。4 μM MT-125使细胞数降至约50%,联合10 Gy照射进一步显著降低细胞数。NAC或ferrostatin 1不仅阻断MT-125细胞毒性,还显著减弱MT-125联合10 Gy照射的效果。最后我们在临床前鼠源Trp53(‒/‒) GEMM模型中考察MT-125联合放疗的治疗效果(图3N)。在该放射抗性模型中,5天每日2 Gy照射对生存无影响,而MT-125单药(每日10 mg/kg)显著延长生存期,复现了早期结果(图2F、2G)。MT-125联合放疗进一步延长生存期,其效果超过放疗与MT-125单药效应的总和。
6. NMIIA通过ROS调控PDGFR信号传导
NMIIA缺失会抑制致癌激酶信号传导,这提示抑制NMII应增强受体酪氨酸激酶(RTK)信号。为验证此假设,我们用5 μM MT-125处理Trp53(‒/‒)细胞48小时。MT-125使PDGFRα及pY849 PDGFRα的表达量增加2-3倍(图4A、4C)。NMIIA缺失也使PDGFRα表达升高7倍(图4D),证实该效应非脱靶作用。MT-125或NMIIA缺失还使AKT(S473)、哺乳动物雷帕霉素靶蛋白(mTOR)(T2446)和S6激酶(T389)的磷酸化水平升高2-4倍(图4E–4I)。由于mTOR是蛋白质翻译的核心调控因子,我们推测MT-125激活mTOR可能促进PDGFRα生成。为验证此点,我们用MT-125联合两种mTOR抑制剂(TOR复合体1/2抑制剂AZD8055和TORC1抑制剂依维莫司)处理Trp53(‒/‒)细胞。两种抑制剂均阻止MT-125诱导的PDGFRα表达增加(图4J)。为探究mTOR激活的作用机制,我们构建了在Rpl22核糖体亚基上表达RiboTag表位的Trp53(‒/‒) GBM细胞系,并用溶剂或5 μM MT-125处理。通过抗HA pull-down分离多聚核糖体后,采用qRT-PCR检测编码PDGFRα的总mRNA及多聚核糖体结合mRNA水平。结果显示(图4K),MT-125特异性使PDGFRα的多聚核糖体mRNA增加近50%(p = 0.0034,双尾t检验)。
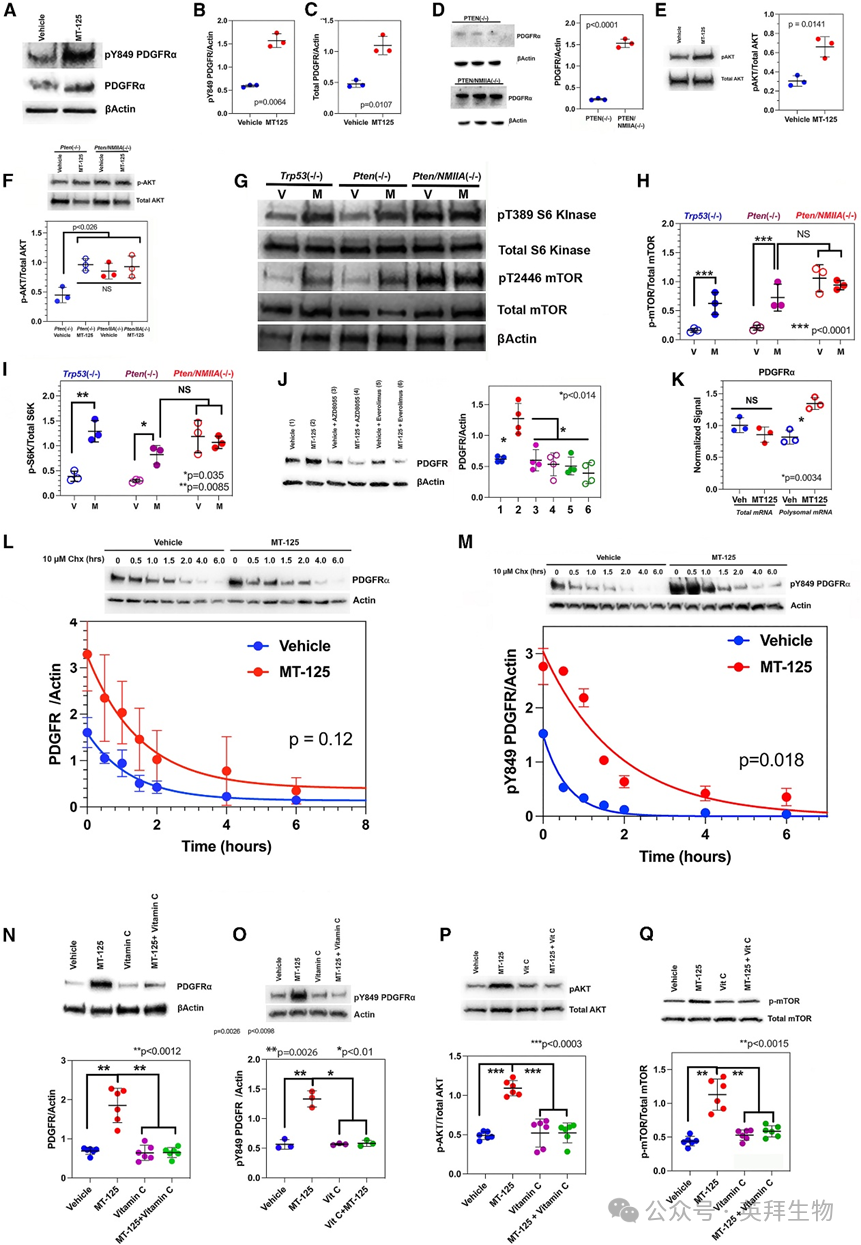
图4 NMII通过ROS调控PDGFR信号传导
有研究报道NMIIA可与PI3K的α亚基(PIK3CA)结合并抑制其活性,该作用可被NMIIA抑制逆转7,这或许能解释MT-125增强AKT、mTOR和S6K激活的现象(图4E–4I)。为验证此机制,我们用抗NMIIA抗体对Trp53(‒/‒) GBM细胞裂解液进行免疫沉淀,并通过液相色谱-串联质谱(LC/MS-MS)分析沉淀物。蛋白质组学分析未检测到NMIIA-PIK3CA结合的证据。第二种可能机制与NMII在受体介导的内吞作用中的已知功能相关——其通过后续细胞内运输将受体运至溶酶体降解35。为验证此点,我们用溶剂或MT-125处理Trp53(‒/‒)鼠源GBM细胞,并添加10 μM cycloheximide阻断蛋白质合成后检测PDGFRα表达随时间的变化。对溶剂(图4L蓝色)和MT-125(图4L红色)数据进行单指数衰减拟合(图4L实线曲线)显示,两条曲线无统计学显著差异(p = 0.12;数据对数变换后双尾t检验),表明MT-125未显著延长总PDGFRα的半衰期。然而,第三种机制将MT-125对线粒体功能的影响与信号传导相联系:除影响DNA和细胞膜完整性外,ROS通过氧化灭活蛋白酪氨酸磷酸酶中两个反应性半胱氨酸,激活RTK依赖性信号传导,从而延长磷酸化激活态RTK的存续时间。我们重复cycloheximide周转实验并检测激活态pY849 PDGFRα,发现MT-125使pY849 PDGFRα的周转速率降低3倍以上(图4M;p = 0.018,数据对数变换后双尾t检验)。若MT-125诱导的PDGFRα信号激活依赖于ROS,则可预测MT-125的信号效应(包括PDGFRα上调和PDGFRα、AKT及mTOR的激活磷酸化)应可被ROS清除剂(如L-抗坏血酸维生素C)逆转。图4N–4Q结果证实了这一预测。但另一种可能是维生素C作为还原剂通过化学反应修饰并灭活MT-125。为排除此干扰,我们在细胞培养基中37℃培养箱内孵育MT-125(含/不含维生素C)。MT-125在3天孵育期间降解极微,无论是否存在维生素C均有>90%化合物保持完整。
7.NMIIA调控自身表达及MAPK依赖性信号传导
由于NMII沿PDGFR-PI3K-AKT-mTOR信号轴调控活性,我们检测了NMIIA或NMIIB表达是否受mTOR调控。用三种mTOR抑制剂(AZD8055、依维莫司和帕沙利西)及PDGFR抑制剂舒尼替尼处理鼠源和人源GBM细胞系,Trp53(‒/‒)和Pten(‒/‒) GEMM细胞系结果如图5A、5B所示。直接或通过PDGFR间接抑制mTOR使NMIIA含量降低约2倍,但不影响NMIIB。若NMII抑制RTK信号而mTOR激活增加NMIIA,则MT-125通过间接激活mTOR也应增强NMII表达。与此一致,我们发现(图5C)MT-125使NMIIA蛋白水平升高约60%。利用RiboTag Trp53(‒/‒) GBM细胞系检测MT-125是否改变编码NMIIA和IIB(分别为Myh9和Myh10)的核糖体结合mRNA水平。MT-125使NMIIA多聚核糖体mRNA增加约50%,但对NMIIB mRNA无影响(图5D、5E)。
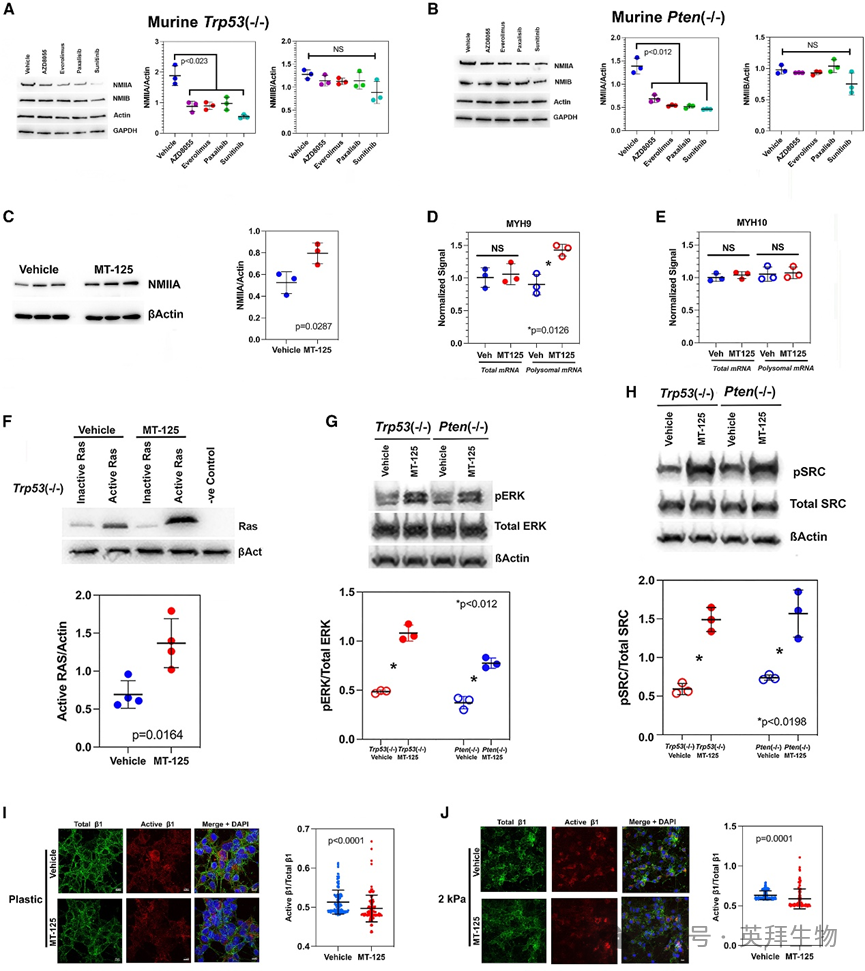
图5 NMIIA表达受mTOR调控并调控MAPK通路
RAS和SRC激活丝裂原活化蛋白激酶(MAPK)通路,并可被RTK活性激活。因此我们检测MT-125是否刺激MAPK信号。MT-125处理Trp53(‒/‒)细胞系使激活态RAS含量翻倍(图5F),相同处理使Trp53(‒/‒)和Pten(‒/‒)细胞的ERK1/2(图5G)和SRC(图5H)磷酸化水平增加2-3倍。我们在5种低传代人源GBM细胞系中也检测到MT-125增强PDGFRα、AKT、mTOR、ERK1/2和SRC的激活。
MAPK通路也受整合素调控——整合素作为牵引力敏感的机械感受器,受NMII产生的力调控。NMII可通过诱导活性构象变化激活整合素,但也可在较高作用力下使整合素与细胞外基质(ECM)脱离连接从而灭活之。抑制NMIIA和IIB介导的整合素解离时,MT-125可能激活整合素信号,进而激活MAPK通路。为此我们在两种层粘连蛋白包被的基底上培养Trp53(‒/‒)细胞:组织培养塑料和具有2 kPa刚度(与宏观脑组织刚度相近)的聚二甲基硅氧烷(PDMS)膜。用溶剂或5 μM MT-125处理这些细胞48小时,并用两种小鼠特异性单克隆抗体染色:一种识别活性β1整合素亚基表位(克隆9EG7,BD Biosciences),另一种识别总β1整合素(克隆HMβ1-1,Biolegend)。硬质(塑料)和软质(2 kPa PDMS)基底上抗活性β1/总β1整合素的荧光强度比值分别见图5I和5J(右侧),显示MT-125降低硬质和软质表面上活性β1整合素的比例(p < 0.0001,双尾t检验),这与NMII在黏着斑成熟中的作用一致。该发现排除了整合素在MT-125激活MAPK信号中的显著贡献。
8. MT-125与致癌激酶抑制剂在体外产生协同效应
我们整合研究结果构建了一个模型:NMII参与两条关键致癌信号通路(图6A)。MT-125通过ROS效应间接增强PDGFR信号传导,进而激活mTOR。由于mTOR上调PDGFRα表达,这将形成正反馈循环——mTOR上调增强PDGFRα表达,进一步强化PI3K活性。但mTOR通过调控NMIIA表达为该循环提供制动机制。MT-125通过解除该制动,同时增强RTK-PI3K-AKT-mTOR和MAPK信号传导。这两条通路通过至少两个层面的激活相互作用连接:一方面是PDGFRα、SRC和RAS之间的相互作用,另一方面是ERK1/2与mTOR之间的相互作用。与此一致,我们发现用PDGFR抑制剂舒尼替尼处理Trp53(‒/‒)细胞可降低AKT磷酸化、mTOR磷酸化、RAS和SRC活性(图6B–6E)。此外,用ERK1/2抑制剂SCH772984(图6F)处理这些细胞可降低mTOR磷酸化水平,即使在MT-125存在下亦然(图6G)。
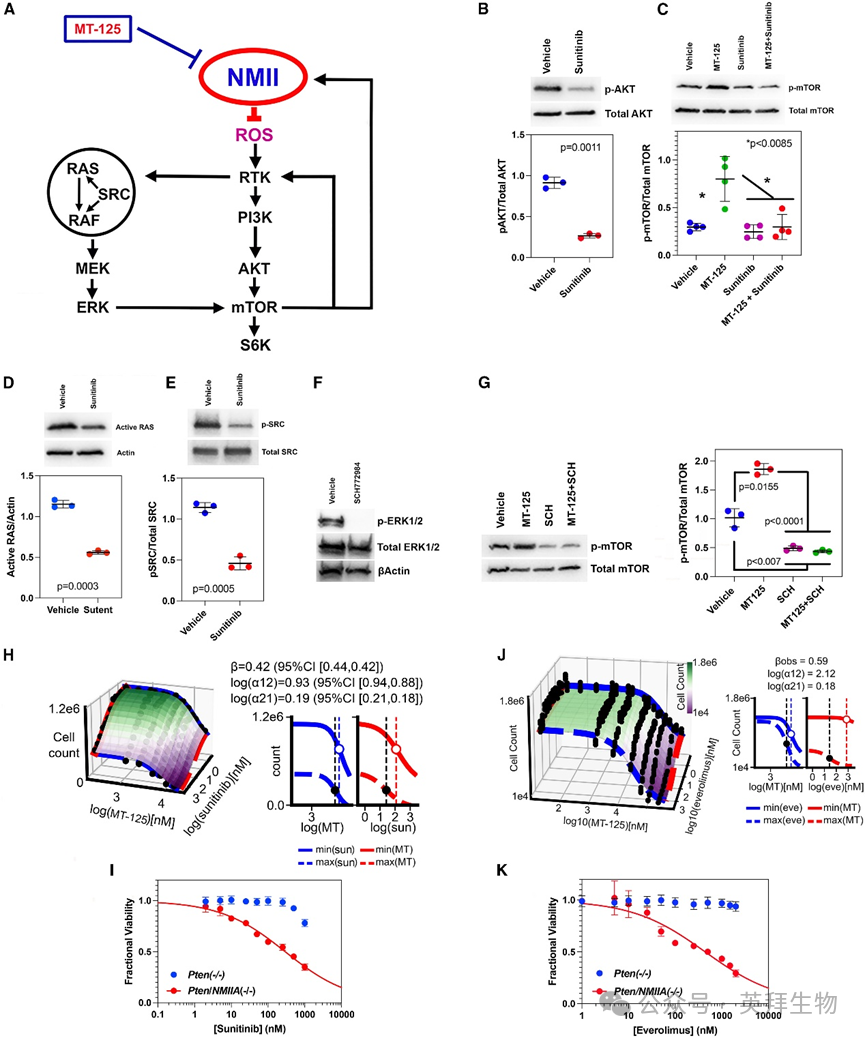
图6 MT-125在体外与致癌信号抑制剂产生协同作用
肿瘤细胞可能对其上调的致癌通路产生依赖性(即"癌基因成瘾"现象),这提示MT-125与RTK-PI3K-AKT-mTOR信号抑制剂联用可能更有效。我们通过体外实验验证:用MT-125+舒尼替尼及MT-125+依维莫司联合处理Trp53(‒/‒) GBM细胞,96小时后检测细胞数量。采用MuSyC算法(可解析效能与效价协同作用)分析结果。该算法通过拟合药物组合数据的剂量反应曲面,计算协同效能程度(β)及协同效价程度(log[α12]和log[α21])。MT-125使舒尼替尼效价提高1.6倍(图6H;log[α21] = 0.19),依维莫司效价提高1.5倍(图6J;log[α21] = 0.18);而舒尼替尼使MT-125效价提高8.5倍(图6H;log[α12] = 0.93),依维莫司使其提高132倍(图6I;log[α21] = 2.12)。剂量反应曲面中的洋红色区域表示观察到协同作用的药物组合。与单药相比,MT-125与舒尼替尼或依维莫司联用分别使细胞数量减少42%(β = 0.42)和59%(β = 0.59)。此外,基因敲除NMIIA也使Pten(‒/‒) GBM细胞对舒尼替尼(图6I)和依维莫司(图6K)更敏感。最后我们筛选MT-125与5种低传代人源GBM细胞系的协同作用:用溶剂、MT-125(5 μM)、舒尼替尼(400 nM)或联合处理48小时,通过CellTiter Glo检测细胞活力。所有细胞系均表达PDGFRα。舒尼替尼与MT-125联用使所有细胞系的活力降低近2倍,效果显著优于单药(p < 0.0001)。
9. MT-125与致癌激酶抑制剂在体内产生协同效应
我们诱导Trp53缺失的GEMM模型,随机分组小鼠接受溶剂(DMSO,每日腹腔注射)、10 mg/kg MT-125(每日腹腔注射)、40 mg/kg舒尼替尼(每周5天口服灌胃)或舒尼替尼+MT-125联合治疗(7天后开始)。治疗4周后处死小鼠,取反转录病毒注射水平的冠状脑切片进行H&E染色和抗HA抗体染色以观察分泌PDGF的肿瘤细胞。与溶剂处理(图7A)相比,MT-125处理组(图7B)的GBM体积显著缩小。如我们既往报道53,舒尼替尼治疗(图7C)增强肿瘤播散,在对侧半球深部出现PDGF阳性肿瘤细胞巢(图7D),可见于铅笔纤维束内(黑色箭头)。而舒尼替尼+MT-125联合治疗(图7E)显著缩小肿瘤体积,仅存孤立HA阳性细胞巢(图7E黑色箭头,图7F放大显示)。
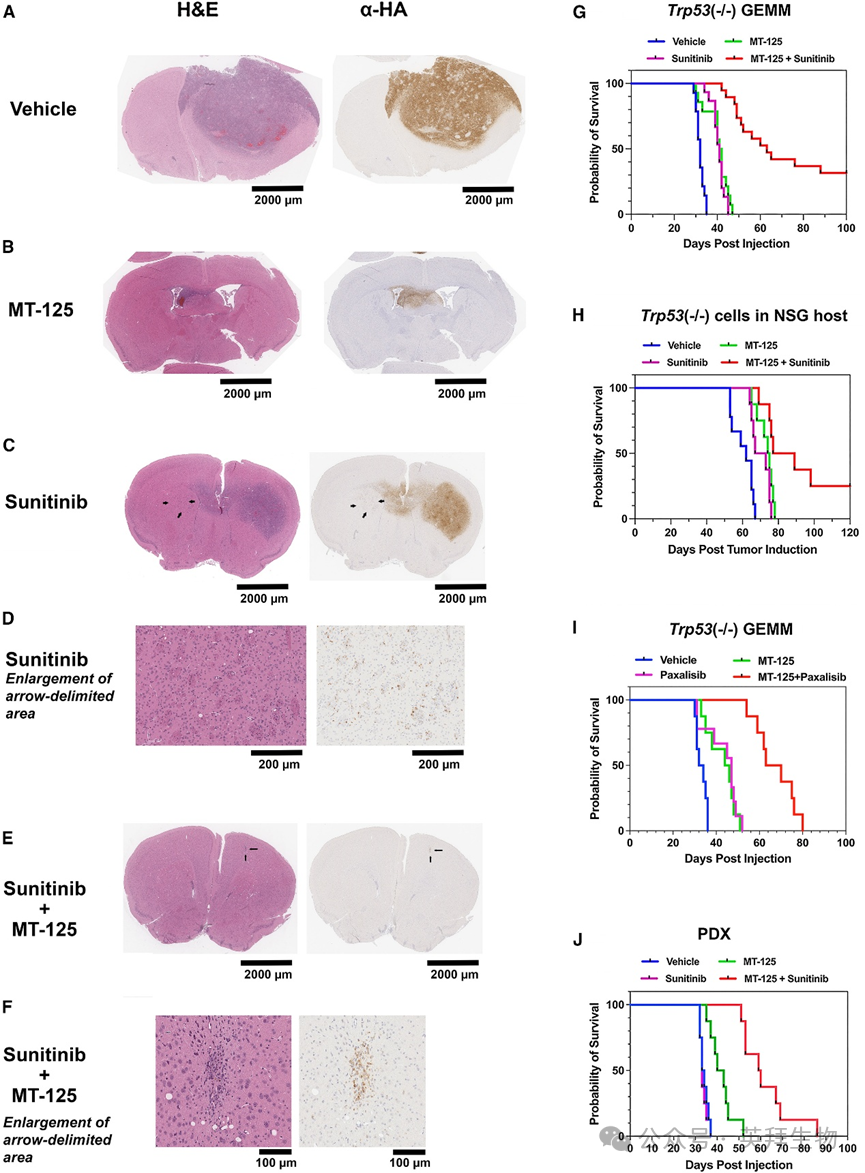
图7 MT-125在体内与致癌信号抑制剂产生协同作用
以相同方式处理第二批小鼠并观察生存期。舒尼替尼(图7G洋红色)或MT-125(图7G绿色)单药治疗较溶剂组(图7G蓝色)延长中位生存期约33%(p < 0.0001,对数秩检验),但所有动物最终均死于肿瘤。而两药联合(图7G红色)使中位生存期较溶剂组延长2倍(p < 0.0001,对数秩检验),并在约40%小鼠中产生长期缓解。MT-125+舒尼替尼给药至第85天停止,该组后续无死亡事件。为评估联合治疗的生存获益在多大程度上依赖淋巴细胞,我们从图7G所用Trp53(‒/‒) GEMM中建立原代鼠源Trp53缺失GBM细胞系,将其原位接种至NSG小鼠,并按图7G方案处理。舒尼替尼(图7H洋红色)或MT-125(图7H绿色)单药治疗较溶剂组(图7H蓝色)延长中位生存期(MT-125 vs溶剂p = 0.002;舒尼替尼vs溶剂p = 0.006,对数秩检验)。但MT-125与舒尼替尼联合(图7H红色)仍比单药更有效(p < 0.026,对数秩检验),并在约25%小鼠中产生长期缓解。根据图6A模型预测,抑制PDGFRα下游致癌激酶的药物与MT-125联用也应增强生存获益。我们在Trp53(‒/‒) GEMM中使用帕沙利西(一种可透过血脑屏障的PI3K/mTOR双重抑制剂)验证该预测。帕沙利西+MT-125联合治疗较单药延长中位生存期(图7I;p < 0.0001,对数秩检验)。最后我们在患者来源异种移植(PDX)模型中重复该治疗方案:给NSG小鼠原位接种50,000个L1人源GBM细胞(图7J),接种后第7天开始治疗。舒尼替尼单药对中位生存期无影响(p = 0.75,对数秩检验),MT-125单药较溶剂组延长生存期25%。而MT-125+舒尼替尼联合治疗较溶剂组延长生存期78%(p < 0.0001),较MT-125单药延长44%(p < 0.0001,对数秩检验)。
参考文献:
Kenchappa RS, Radnai L, Young EJ, Zarco N, Lin L, Dovas A, Meyer CT, Haddock A, Hall A, Toth K, Canoll P, Nagaiah NKH, Rumbaugh G, Cameron MD, Kamenecka TM, Griffin PR, Miller CA, Rosenfeld SS. MT-125 inhibits non-muscle myosin IIA and IIB and prolongs survival in glioblastoma. Cell. 2025 Aug 21;188(17):4622-4639.e19. doi: 10.1016/j.cell.2025.05.019. Epub 2025 Jun 10. PMID: 40499543; PMCID: PMC12354127.