






CLDN4棕榈酰化促进肝细胞癌向胆管谱系转化及仑伐替尼耐药
肝细胞癌(HCC)具有显著的可塑性,能够通过表型转换促进药物耐受状态并规避药物诱导的细胞毒性。本研究发现,与紧密连接蛋白Claudin 4(CLDN4)相关的肝向胆管谱系转化(HBT)是缓解HCC仑伐替尼耐药潜在靶点。CLDN4在仑伐替尼耐药患者中表达更为普遍。CLDN4半胱氨酸残基C104和C107位的棕榈酰化修饰可调控赖氨酸残基K103位的泛素化,抑制网格蛋白介导的内吞作用,并维持CLDN4在脂筏中的锚定。锚定状态的CLDN4通过驱动接触蛋白-1向脂筏募集并激活Notch信号通路,促进HCC细胞发生表型转化,从而增强对仑伐替尼的耐药性。研究证实CLDN4抑制剂丹酚酸B能同时逆转HCC的HBT进程和仑伐替尼耐药性。此外,联合化疗对发生HBT的HCC患者展现出显著治疗效果。该研究于2025年7月发表在《Cell Reports Medicine》,IF:10.6。
技术路线:
主要研究结果:
1.CLDN4可作为缓解肝细胞癌仑伐替尼耐药的潜在靶点
为探究仑伐替尼耐药的关键分子机制,我们通过体内诱导法建立自发性HCC模型并获取潜在耐药细胞:通过持续仑伐替尼暴露诱导HCC小鼠产生药物耐受性,经体内外实验验证原代HCC细胞的药物耐受性增强(图1A-1E)。收集耐受仑伐替尼的原代细胞进行蛋白质组学分析,同时采集3对仑伐替尼耐药/敏感患者的肿瘤样本进行单细胞测序(Fu-LR Sc队列)。CLDN4在耐药患者中表达上调最显著,且是仅有的在耐药细胞系和患者中同步上调的分子(图1F-1H)。经严格质控后保留93,776个细胞进行单细胞RNA测序(scRNA-seq),对基因表达数据标准化后,采用主成分分析和均匀流形近似与投影(UMAP)进行降维聚类,再通过拷贝数变异分析区分恶性与非恶性细胞。根据标志基因将细胞群分为9类:上皮/肿瘤细胞(24,463个,26.1%,标志基因EPCAM/ALDH1A1/ALB)、B细胞(4,341个,4.6%,MS4A1/CD79A)、T细胞(36,264个,38.7%,CD3D/CD3E)、自然杀伤细胞(2,168个,2.3%,FGFBP2)、中性粒细胞(2,459个,2.6%,ITGAM/CD33)、巨噬细胞(12,624个,13.5%,CD68/CD163/CD14)、单核细胞(5,898个,6.3%,ITGAX)、内皮细胞(4,120个,4.4%,PECAM1)及平滑肌细胞(1,439个,1.5%,PECAM1/VWF)。值得注意的是,CLDN4定位于恶性细胞(图1I和S1A)。既往研究评估多株HCC细胞系对仑伐替尼的敏感性,将SUN449、JHH1、Huh6等列为耐药系,SUN398、Huh7等列为敏感系。本研究发现CLDN4在多数耐药系中表达显著高于敏感系(图1J、1K)。为进一步验证CLDN4在耐药中的作用,收集55例术前接受仑伐替尼治疗患者的残余肿瘤组织(Fu-LR队列)及55例直接手术患者的未治疗肿瘤组织。免疫组化显示CLDN4定位于肿瘤细胞膜(图1L、1M),且残余队列中CLDN4阳性或疾病稳定患者比例显著高于未治疗队列(图1N)。CLDN4高表达患者术后预后较差(图1O;表S1)。通过采集12例患者治疗前活检样本发现,仑伐替尼应答组(n=4)肿瘤组织的CLDN4免疫荧光强度显著低于耐药组(n=8)(图1P)。这些结果表明CLDN4可作为克服HCC仑伐替尼耐药的潜在治疗靶点。
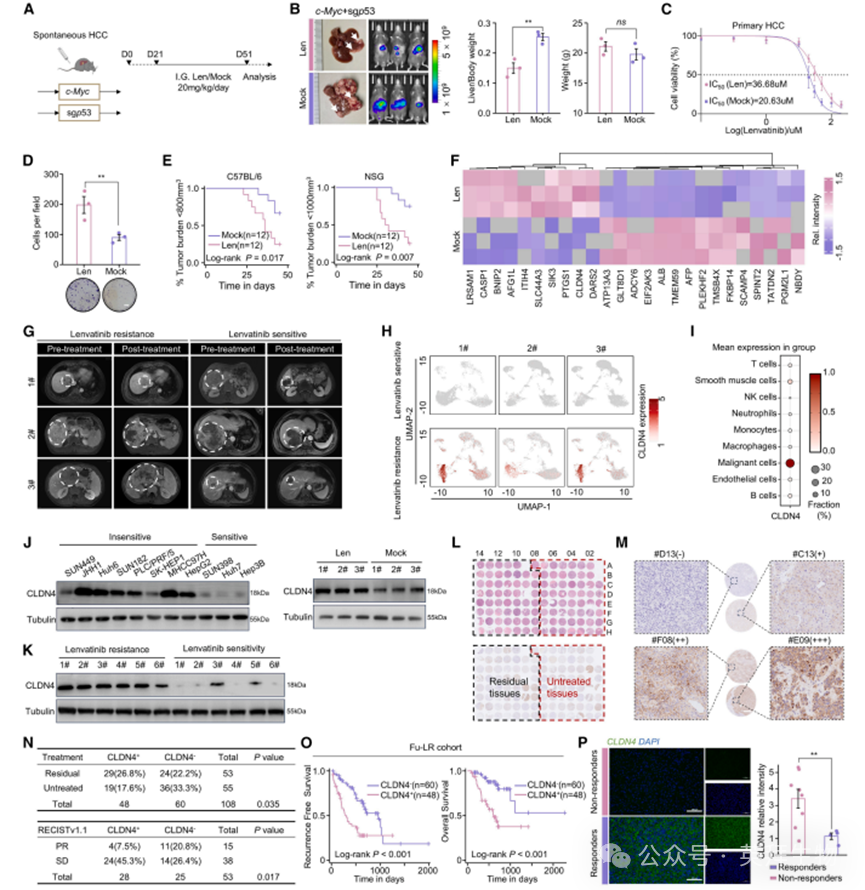
图1.CLDN4可作为缓解肝细胞癌仑伐替尼耐药的潜在靶点
2.CLDN4高表达与HCC胆管谱系转化及仑伐替尼耐药相关
为探究CLDN4在HCC仑伐替尼耐药中的特征,我们整合Fu-LR Sc队列数据集及两项已发表研究(图S1B、S1C)。经过严格质控过滤后,对各数据集进行无监督聚类以区分恶性细胞,保留高表达典型恶性细胞标志基因的簇群。最终对scRNA-seq分析中56例患者的69,711个恶性细胞进行分类,聚类为12个不同亚群(图2A)。所有亚群均呈现不同水平的肝系/胆系标志物特征(通过HCC与ICC差异表达谱确定)。值得注意的是,CLDN4表达与胆系标志物呈正相关,而与肝系标志物呈负相关(图2B、2C、S1D)。为阐明恶性细胞亚型间的谱系转化与分化潜能,采用Monocle 2和CytoTRACE算法推导伪时间轨迹和分化水平(图2D-2F)。随着CLDN4表达升高,胆系标志物(EPCAM、SOX9、CEACAM1等)逐渐上调,而肝系标志物(载脂蛋白E、白蛋白、纤维蛋白原γ/α链等)相应下调,证实HCC患者存在胆管谱系转化(图2G、2H)。基因集富集分析显示,CLDN4高表达患者的基因特征与胆管癌(CHOL)基因集呈正相关,与肝细胞癌(LIHC)基因集呈负相关(图2I、S1E、S1F)。qPCR分析证实小鼠CLDN4表达在胚胎16天达峰(图2J),从发育早期至成熟肝细胞逐渐下降(图2K)。在患者组织中,CLDN4表达从远端正常肝组织→癌旁组织→HCC组织逐级升高,仑伐替尼耐药肿瘤组织中表达进一步上调(图2L)。值得注意的是,CLDN4水平与肿瘤病理分级呈正相关(图2M)。过表达CLDN4可上调所有胆系标志物并抑制肝系标志物表达(图S1G、S1H)。此外,敲低CLDN4能显著降低HCC细胞在体内外对仑伐替尼的耐药性(图2N-2P、S1I)。这些发现表明CLDN4过表达通过促进HCC胆管谱系转化介导仑伐替尼耐药。
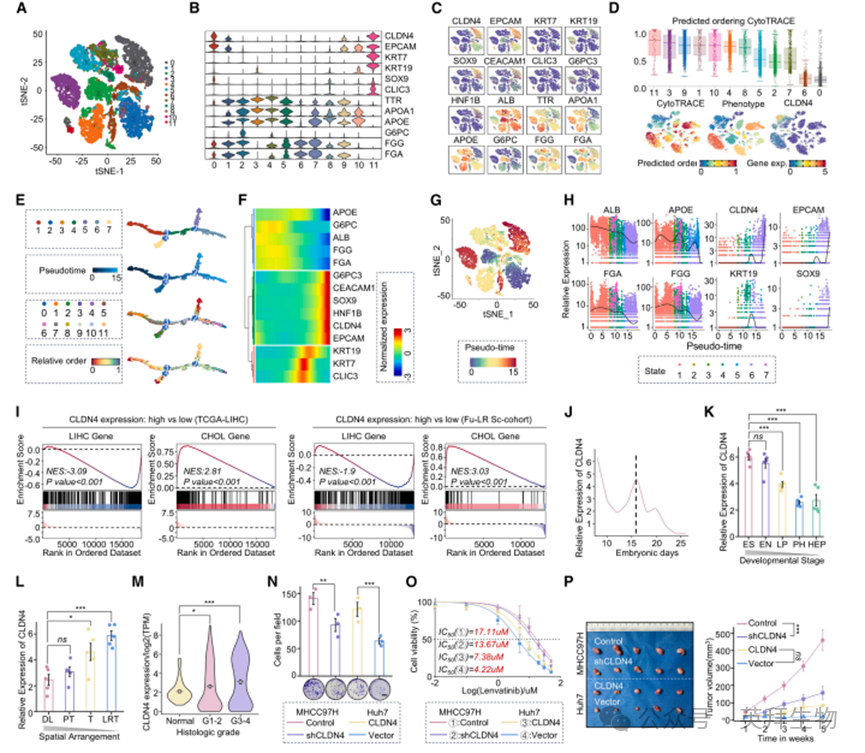
图2.CLDN4高表达与HCC胆管谱系转化及仑伐替尼耐药相关
3.棕榈酰基转移酶zDHHC5介导CLDN4在进化保守半胱氨酸残基C104和C107位的S-棕榈酰化修饰
既往研究表明脂质修饰可能参与claudin蛋白的信号转导和紧密连接组装。例如,与非棕榈酰化突变体相比,棕榈酰化CLDN蛋白能更有效地分布到脂质双分子层中。为探究CLDN4的脂质修饰,我们提取HCC细胞系膜组分进行棕榈酰化蛋白质组测序(图3A-3C)。通过点击化学反应连接生物素-叠氮化物和酰基-生物素交换(ABE)两种方法证实HCC中存在棕榈酰化修饰(图3D、S2A、S2B)。有趣的是,使用棕榈酰化抑制剂2-BP处理后,两种HCC细胞系质膜上的CLDN4表达显著降低(图3E、S2C)。相反,通过酰基蛋白硫酯酶(APT)选择性抑制剂palmostatin B和ML348增强棕榈酰化后,MHCC97H细胞中CLDN4表达增加(图S2D、S2E)。序列比对显示CLDN4的C104、C107、C182、C183和C185位点在不同物种间保守(图3F、3G)。我们采用基于模组的预测工具CSS-palm 4.0预测显示,含C182、C183和C185的CLDN4 C端区域具有较高棕榈酰化概率。但通过ABE-质谱分析证实,HCC中CLDN4的实际棕榈酰化位点位于跨膜区(TR)的C104和C107位点(图3H、3I、S2F-S2H)。这凸显了算法预测与实际修饰位点之间的差异。
蛋白质棕榈酰化通常由特异性棕榈酰转移酶介导,其选择性取决于酶的底物特异性、亚细胞定位、调控信号及底物结构特征。例如,位于不同细胞区室的棕榈酰转移酶仅修饰与其发生物理相互作用的底物。在HCC中,zDHHC5/7/9/16/17/18/20/21的高表达与不良预后相关。为鉴定MHCC97H细胞中CLDN4的主要棕榈酰转移酶,我们在HEK293细胞中分别共表达HA标记的zDHHC蛋白与FLAG标记的CLDN4。免疫共沉淀结合免疫印迹实验表明zDHHC1/5/20/21/24可与CLDN4结合(图S3A)。随后发现敲低zDHHC5会导致内源性CLDN4棕榈酰化水平下降(图S3B)。因此我们推测zDHHC5参与CLDN4的动态棕榈酰化,并利用AlphaFold3进行分子对接建模(图3J)。ABE实验证实zDHHC5促进CLDN4在C104和C107位点的棕榈酰化(图3K-3N、S3C-S3E)。在Fu-LR队列中,zDHHC5高表达与CLDN4高表达及HCC患者不良预后相关(图3O、3P、S3F、S3G)。zDHHC5在仑伐替尼耐药患者和细胞系中也显著上调(图3Q、S3H、S3I)。敲低zDHHC5后,HCC细胞对仑伐替尼的耐药性在体外显著降低,反之亦然(图S4A-S4H)。这些结果表明zDHHC5介导CLDN4在进化保守位点C104和C107的S-棕榈酰化修饰。
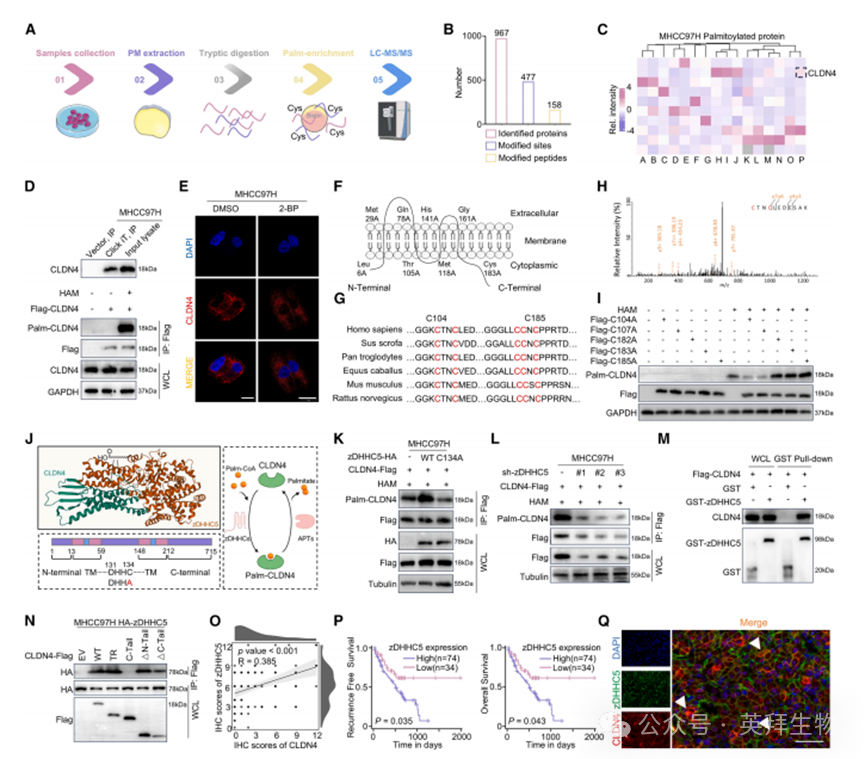
图3.棕榈酰基转移酶zDHHC5介导CLDN4在进化保守半胱氨酸残基C104和C107位的S-棕榈酰化修饰
4.CLDN4棕榈酰化是其定位于脂筏的必要条件
Claudin蛋白通常定位于脂筏等功能性膜结构域发挥作用。我们探究棕榈酰化修饰是否能促进CLDN4锚定于脂筏。在2-BP处理或敲低zDHHC5的MHCC97H细胞中,脂筏上的CLDN4水平显著降低,该现象可被内吞抑制剂Dynasore和溶酶体抑制剂氯喹(CQ)逆转,但蛋白酶体抑制剂MG132无此效应(图4A、4B)。对CLDN4免疫沉淀物的质谱分析提示其与网格蛋白相互作用蛋白1(CLINT1)及溶酶体相关膜蛋白2(LAMP2)存在相互作用(图S6A)。由此我们推测CLDN4通过网格蛋白介导的内吞(CME)途径转运至溶酶体降解。实验验证了CLDN4与网格蛋白重链(CLTC)及LAMP2的结合(图S6B)。值得注意的是,我们发现CLDN4与CLINT1存在直接相互作用(图4C-4E、S6C、S6D)。正如预期,敲低CLINT1可有效挽救去棕榈酰化诱导的CLDN4降解(图4F),而蛋白酶体抑制剂MG132未能延缓CLDN4蛋白降解速率(图S6E)。免疫荧光显示2-BP和shzDHHC5处理细胞中CLDN4与CLTC/LAMP2共定位显著增加,shCLINT1处理细胞中则明显减少(图4G、4H)。但目前尚未阐明为何CLDN4棕榈酰化能影响CME过程——该过程已知受动态泛素化与去泛素化调控。另有研究表明棕榈酰基可阻断泛素结合,提示这两种修饰在分子水平存在交叉对话与竞争。实验结果表明抑制棕榈酰化显著增强CLDN4与泛素或CLINT1的相互作用(图4I)。为揭示介导CLDN4泛素化的E3泛素连接酶,我们从CLDN4免疫沉淀物中筛选出三重基序蛋白33(TRIM33)。值得注意的是,敲低TRIM33可升高MHCC97H细胞中CLDN4蛋白水平并降低其泛素化积累(图4J-4M),而MG132不影响CLDN4结合的泛素水平(图S6F)。此外,基于BDM-PUB(贝叶斯判别法预测蛋白泛素化位点工具,预测结果,K103位点可能是受棕榈酰化影响的潜在单泛素化位点。K48连接泛素化通常通过蛋白酶体途径介导降解,而K63连接泛素化介导内吞和DNA损伤修复等过程。我们在MHCC97H细胞中同时转染HA标记的K63和FLAG标记的K103突变型CLDN4,结果表明CLDN4 K103位点的泛素化由K63连接泛素链介导(图4N、4O)。综上所述,CLDN4半胱氨酸残基C104和C107位的棕榈酰化修饰促进其锚定于脂筏(图4P)。
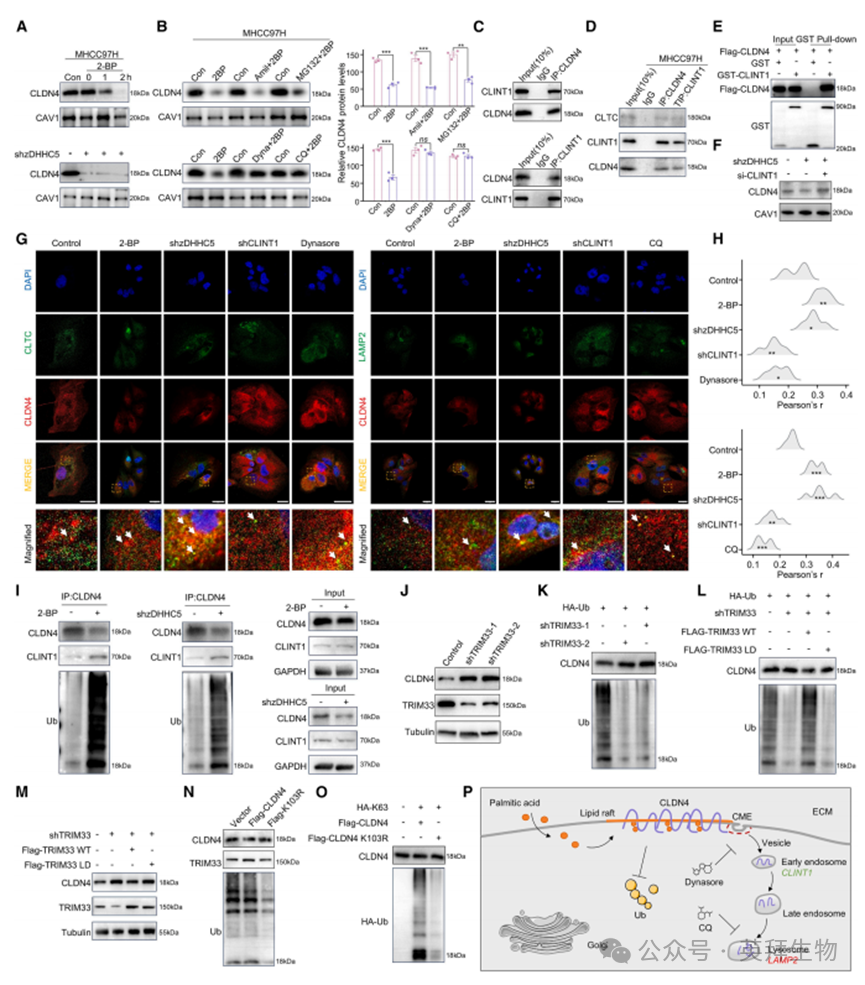
图4. CLDN4棕榈酰化是其定位于脂筏的必要条件
5.CLDN4通过激活Notch通路改变肿瘤细胞可塑性
合成竞争性结合靶分子棕榈酰化区域的肽段是开发特异性抑制剂的策略之一。我们据此合成针对CLDN4棕榈酰化的竞争性肽CPP-S4,该肽能有效抑制CLDN4的膜稳定性并增强仑伐替尼的细胞毒性效应(图5A-5E、S7A-S7F)。经CPP-S4处理的MHCC97H细胞中,胆管谱系转化(HBT)相关基因表达不同程度下调(图5F、5G、S7G、S7H)。转录组分析显示细胞从仑伐替尼耐药特征向敏感特征转变(图S7I)。值得注意的是,通路富集分析表明Notch相关信号通路受抑制最显著,该结果通过免疫印迹和qPCR得到验证(图5H-5I、S7J)。重要的是,CLDN4过表达引起的HBT标志物升高可被γ-分泌酶抑制剂RO4929097显著逆转(图5J、5K)。我们进一步鉴定发现NICD(Notch胞内结构域)-RBPJ(重组信号结合蛋白Jκ区)结合位点位于HBT标志物的启动子区域(图5L、5M)。染色质免疫沉淀(ChIP)-PCR及ChIP实验证实NICD-RBPJ显著富集于HBT标志物启动子区(图5N、5O)。设计野生型和突变型NICD-RBPJ位点探针进行DNA亲和沉淀实验,结果显示NICD-RBPJ仅与野生型HBT标志物启动子区寡核苷酸结合(图5P)。双荧光素酶报告实验进一步证实NICD-RBPJ是HBT标志物启动子转录活性及基因表达的必要条件(图5Q)。综上所述,这些结果表明CLDN4通过Notch信号通路诱导HBT标志物表达。
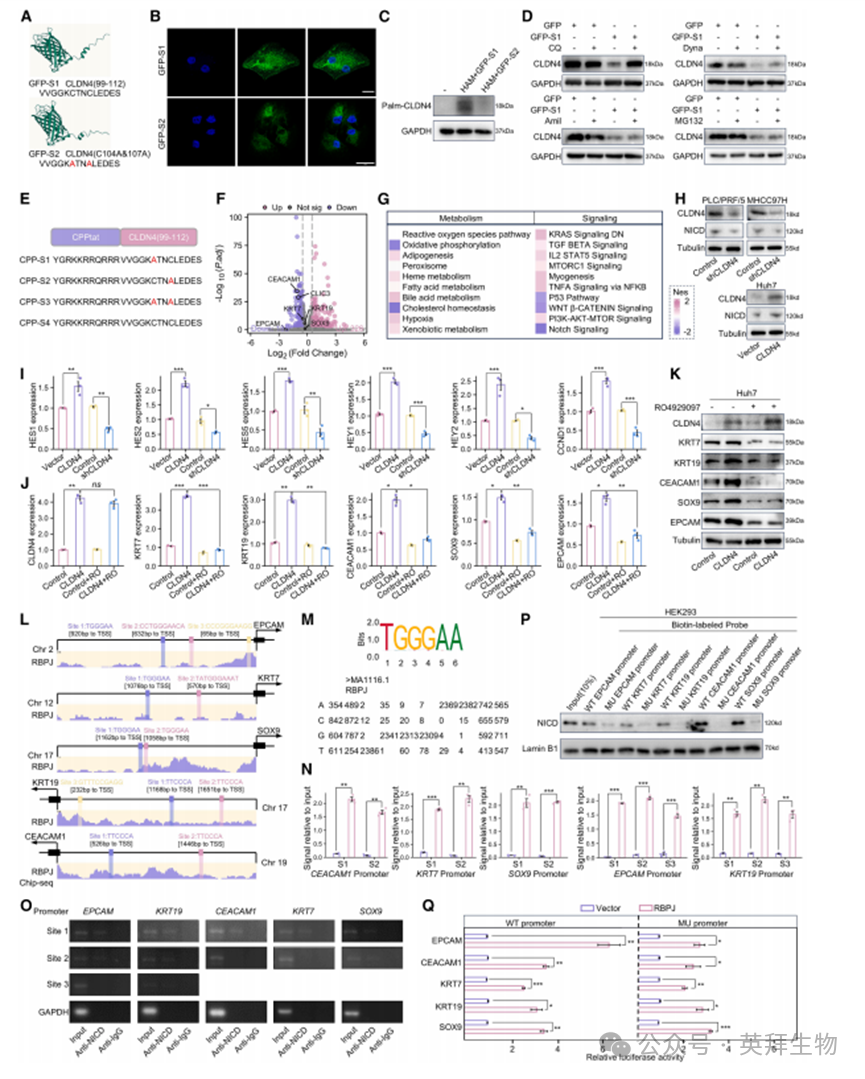
图5. CLDN4通过激活Notch通路改变肿瘤细胞可塑性
6.CLDN4驱动CNTN1向脂筏募集并激活Notch信号通路
通过MHCC97H细胞膜共免疫沉淀实验,我们发现CLDN4与CNTN1而非JAG1存在相互作用(图6A、6B)——这两种分子均被报道可激活Notch信号。免疫荧光共定位及TCGA-LIHC数据表明CNTN1与CLDN4表达具有一致性(图6C、6D)。接下来我们探究CLDN4如何通过结合促进CNTN1活性。CNTN1是一种神经细胞黏附蛋白,由6个N端免疫球蛋白(Ig)结构域和4个纤连蛋白样重复序列组成(图6E)。通过构建带有FLAG标签的纤连蛋白III样和Ig样C2型1突变体,发现CNTN1的纤连蛋白III结构域参与CLDN4-CNTN1相互作用(图6F)。由于CNTN1通过糖基磷脂酰肌醇(GPI)锚定在质膜上,其几乎完全定位於脂筏中(图6G)。因此我们提出假说:棕榈酰化CLDN4通过招募CNTN1至脂筏激活Notch通路。实验结果表明CLDN4不影响CNTN1总表达水平,但显著改变其在脂筏中的分布(图6H、6I)。脂筏抑制剂甲基-β-环糊精(MβCD)显著降低CLDN4与CNTN1在膜上的共定位,并抑制Notch信号传导和胆管谱系转化(图6J-6N)。下调CNTN1或添加MβCD可消除CLDN4过表达HCC细胞中仑伐替尼半抑制浓度(IC50)的变化,反之亦然(图6O)。皮下成瘤实验也证实MβCD或RO4929097能增强仑伐替尼的抗肿瘤效果(图6P)。这些发现强调棕榈酰化CLDN4通过驱动CNTN1向脂筏募集进而放大Notch致癌通路。
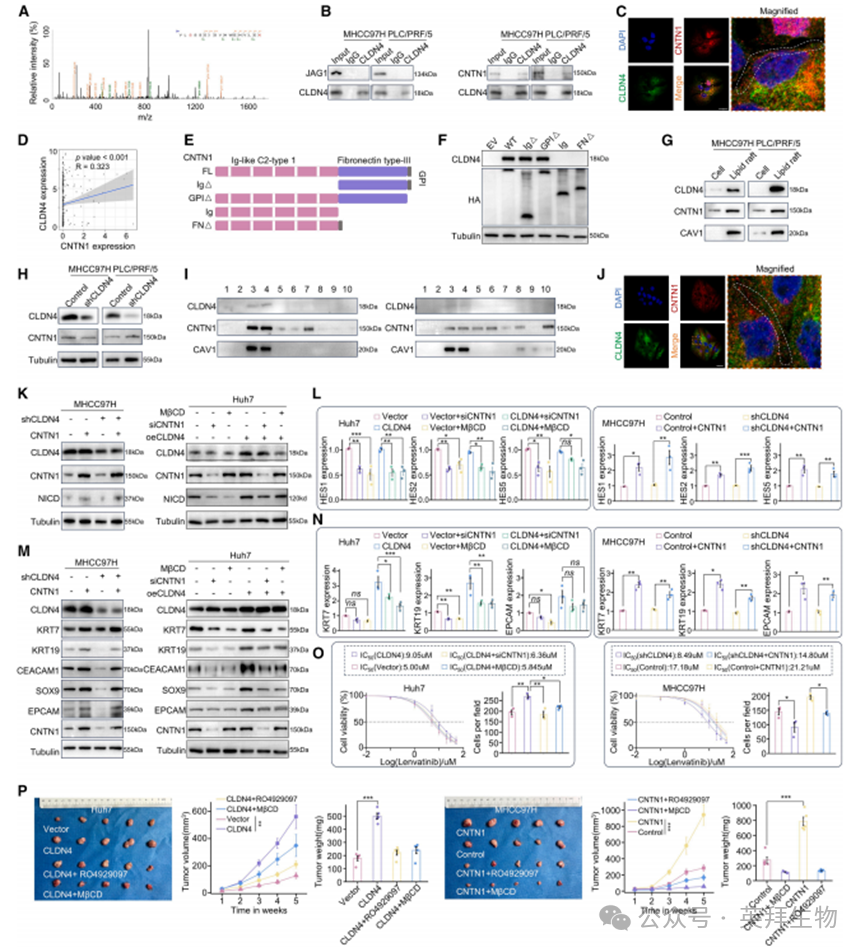
图6. CLDN4驱动CNTN1向脂筏募集并激活Notch信号通路
7.通过虚拟筛选发现CLDN4拮抗剂
我们随后尝试寻找抑制CLDN4的临床适用方法。首先考虑采用靶向CLDN4的单克隆抗体或抗体-药物偶联物(ADC)策略向CLDN4阳性细胞递送细胞毒性药物——该策略已应用于其他claudin蛋白靶点。但由于CLDN4在食管和胃黏膜等正常上皮组织中高表达,这两种方法均不适用于靶向仑伐替尼耐药HCC,直接靶向杀伤会导致严重胃肠道副作用。靶向CLDN4的棕榈酰化修饰可能是更有效的策略。我们收集17681种小分子药物(Topscience,T001),对CLDN4-zDHHC5结合域三维模型进行分子对接。经过虚拟筛选和毒性评估,确定丹酚酸B(SalB)为高效安全的CLDN4拮抗剂(图7A、7B、S8A-S8J)。为探究SalB对HCC发病机制的影响,我们在Zdhhc5基因敲除(Zdhhc5−/−)小鼠中建立自发性HCC模型(图S9A)。与野生型(WT)相比,Zdhhc5−/−小鼠自发性肿瘤进展更慢(肝重/体重比降低),肝损伤更轻(ALT/AST值下降)(图S9B)。Zdhhc5−/−小鼠HCC组织中CLDN4表达水平及棕榈酰化程度均显著降低(图S9C、S9D)。相应地,SalB能在不引起显著肝毒性的前提下抑制WT小鼠肿瘤生长(图S9E-S9H)。这些结果表明SalB是一种有效的CLDN4拮抗剂。
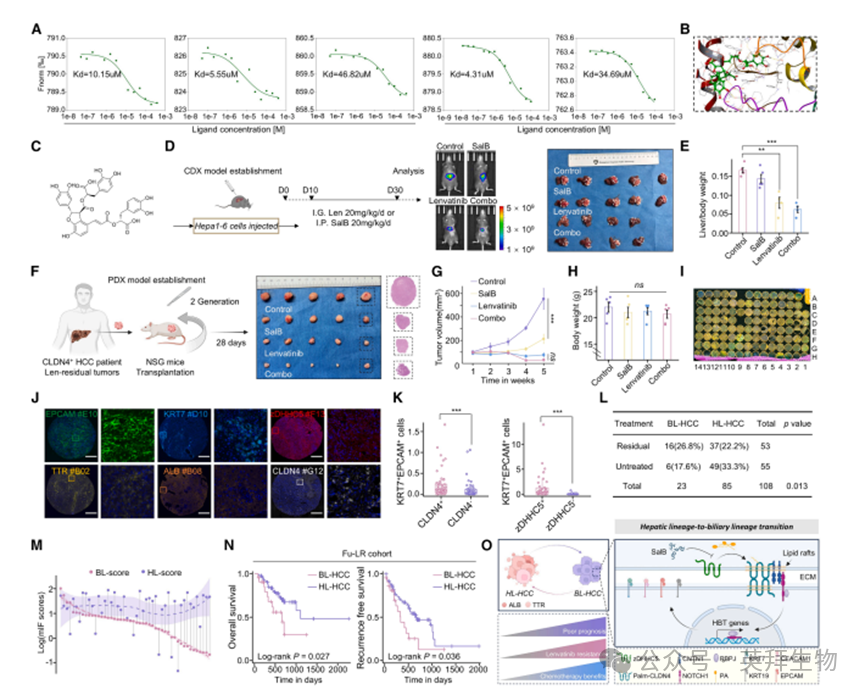
图7. 通过虚拟筛选发现CLDN4拮抗剂
8.CLDN4诱导的胆管谱系转化与HCC不良预后相关
为在体内模型中验证发现,我们通过将Hepa1-6细胞注射至C57BL/6小鼠肝脏建立原位模型。10天后检测到肿瘤形成,并给予仑伐替尼、SalB或对照剂治疗20天。联合治疗组肿瘤体积显著减小(图7C-7E)。构建CLDN4阳性仑伐替尼耐药人源肿瘤异种移植模型(LR-PDXs),SalB显著增强仑伐替尼在LR-PDXs中的抗肿瘤效果且未观察到明显毒性(图7F-7H)。由于HCC患者通常不似胆管癌(ICC)患者接受化疗,混合型肝癌患者是否应接受化疗尚无明确共识。我们探究具有胆管谱系转化(HBT)的HCC患者是否能从化疗中获益。结果表明,尽管伴随小鼠体重显著下降,吉西他滨/顺铂联合仑伐替尼对CLDN4+KRT7+EPCAM+ PDXs模型疗效最佳(图S10A-S10D)。值得注意的是,SalB给药可部分逆转HBT,治疗组HBT标志物表达显著低于对照组(图S10E)。而对于CLDN4−KRT7−EPCAM− PDXs模型,联合化疗未带来显著获益(图S10F-S10J)。
为验证HBT与仑伐替尼耐药的关系,我们对Fu-LR队列进行多重免疫组化(mIHC)并开展类组织流式分析,实现对肿瘤微阵列每个位点单细胞的定量荧光分析。胆管谱系HCC在残余队列中占比显著更高,反之亦然(图7I-7L、S11A、S11B)。此外,肝系与胆系标志物表达模式完全相反,提示HCC中胆管特性与肝细胞特性相互制约并可相互转化(图7M)。与无HBT的HCC病例相比,具HBT特征的患者总生存期和无复发生存期显著缩短(图7N)。综上所述,HBT与HCC仑伐替尼耐药及不良预后相关,同时可能从化疗中获益(图7O)。
结论:
该研究揭示了肝细胞癌(HCC)中紧密连接蛋白4(CLDN4)的棕榈酰化通过促进肝细胞向胆管细胞谱系转化(HBT)和激活Notch信号通路,导致仑伐替尼耐药的分子机制。棕榈酰化位点C104和C107由zDHHC5催化,增强了CLDN4在脂筏中的锚定,抑制了其内吞作用,从而维持其在细胞膜上的稳定表达。棕榈酰化的CLDN4将接触蛋白1(CNTN1)募集到脂筏中,激活Notch信号通路,上调胆管细胞谱系标志物,下调肝细胞谱系标志物,促进HBT。CLDN4的高表达与仑伐替尼耐药性密切相关。Salvianolic acid B(SalB)通过抑制zDHHC5,降低CLDN4的棕榈酰化水平,减少HBT,恢复仑伐替尼的敏感性。联合化疗(如吉西他滨/顺铂)对发生HBT的HCC患者可能有效。
参考文献:
Xu M, Zheng Y, Chen J, Gao C, Zhu M, Ma A, Liang B, Xu W, Fan J, Zhou H, Ke A, Shen Y. CLDN4 palmitoylation promotes hepatic-to-biliary lineage transition and lenvatinib resistance in hepatocellular carcinoma. Cell Rep Med. 2025 Jul 15;6(7):102208.