






活化T细胞通过巨噬细胞再教育打破肿瘤免疫抑制
在这项研究中,作者观察到,在人类和小鼠黑色素瘤中,T细胞活化通过TNFα信号传导减弱了肿瘤相关巨噬细胞(TAM)中造血前列腺素-D2合酶(HPGDS)的转录。从机制上讲,HPGDS通过DP1和DP2激活在TAM中安装前列腺素D2(PGD2)自分泌环,维持其原癌表型,并通过PGD2-DP1轴促进CD8+T细胞的旁分泌抑制。遗传或药理学HPGDS靶向诱导TAMs的抗肿瘤特征,并有利于CD8+T细胞的募集、激活和细胞毒性,从而使肿瘤对αPD1敏感。相反,TAMs中HPGDS的过表达或系统性TNFα阻断维持了肿瘤前环境和αPD1耐药性,阻止T细胞下调HPGDS。一致地,对αPD1耐药的患者和小鼠未能抑制TAM中的HPGDS,这进一步证明绕过HPGDS对于有效治疗αPD1是必要的。总的来说,作者揭示了一种T细胞活化控制先天免疫系统的机制,作者建议靶向HPGDS/PGD2以克服免疫疗法耐药性。该研究于2025年7月发表在《Cancer Discovery》,IF 33.3分。
技术路线:
主要研究结果:
1 HPGDS在黑色素瘤中由TAM亚群表达
为鉴定在ICB治疗中由肿瘤相关巨噬细胞(TAMs)表达并具有临床相关性的花生四烯酸代谢基因,作者分析了一个自建的黑色素瘤患者单细胞 RNA 测序(scRNA-seq)数据集,该数据集包括对免疫检查点阻断(ICB)治疗有应答的患者 [responders (R)] 和无应答的患者 [nonresponders (NR)],采样时间点为治疗前和治疗后 [on-treatment (OT)](23 例患者接受了 αPD1 治疗,其中 6 例同时接受了 αCTLA4 治疗;bioRxiv 2023.12.14.571631)。在所有 TAM 的基因表达分析中,作者发现45个花生四烯酸代谢相关基因中,HPGDS 是仅有的在无应答患者 ICB 治疗 2–3 周后显著上调的基因(图 1A)。
在同一数据集的所有免疫细胞簇中,HPGDS 的表达仅限于一个小的肥大细胞簇和部分巨噬细胞亚群(图 1B)。对两个公开的黑色素瘤患者 scRNA-seq 数据集进行分析后也得出了相同结论,即 HPGDS 表达限制在 TAM 中(图 1C 和 D;refs. 22, 23)。在作者自建的数据集中,HPGDS 特异性表达于一个以高水平表达 LYVE1、MRC1、NRP1 和 CD163 为特征的巨噬细胞簇中,而在其他巨噬细胞亚群(如PLA2G2D_macrophages、SPP1_macrophages、CXCL9_macrophages 和 CCL3_macrophages)以及单核细胞中几乎不表达或完全不表达(图 1E)。这一表达模式在所有患者中保持一致(附图 S1A)。进一步比较所有 TAM 的基因表达模式发现,高表达 HPGDS 的巨噬细胞富集促肿瘤特征基因(如 LYVE1、MRC1、VSIG4、C1QB 和 MS4A4A;图 1F;ref. 24)。在 TCGA 黑色素瘤(皮肤切除黑色素瘤)数据集中(包含 473 名患者),作者同样验证HPGDS与促肿瘤巨噬细胞特征基因之间的相关性(附图 S1B)。
在蛋白水平上,作者对14例患者的样本(包括原发性黑色素瘤、播散性黑色素瘤以及转移性黑色素瘤:转移中转灶、皮肤转移和淋巴结或肝转移)进行了免疫组化(IHC)验证,结果显示大约20%的CD163+ 促肿瘤TAMs与HPGDS 共定位(图 1G 和 H;附图 S1C;附表 S1)。值得注意的是,在ICB应答患者(R/OT)中,HPGDS 在RNA和蛋白水平均被完全下调(同时伴随 CD8+ T 细胞扩增增强;ref. 25)(图 1I 和 J);而在ICB无应答患者(NR/OT)中,HPGDS 表达仍然保持高水平(图 1I 和 J)。在 LYVE1_macrophages 中的177个差异表达基因(DEG)中,只有 HPGDS 和另一个基因在 αPD1 治疗后显著下调(附图 S1D)。在 αPD1 治疗后,其他巨噬细胞亚群中的 HPGDS 表达始终处于低水平或不可检测(附图 S1E)。在配对样本分析中(同一患者治疗前 vs. 治疗后),应答患者的 HPGDS 表达在 ICB 治疗后出现下降,尽管样本量较小(N = 9),但差异几乎达到显著性(P = 0.059;附图 S1F)。
为进一步验证这一观察结果,作者又分析了一个独立的患者队列(46 例黑色素瘤患者,均接受 αPD1 治疗,其中 11 例同时接受 αCTLA4 治疗;ref. 23)。结果同样显示,在应答患者中,巨噬细胞的 HPGDS 在 ICB 治疗后被下调(附图 S1G)。
综上,这些结果提示:HPGDS 可作为一个标志物,用于识别潜在的免疫抑制性巨噬细胞亚群,而其下调则是 αPD1 治疗有效的信号。
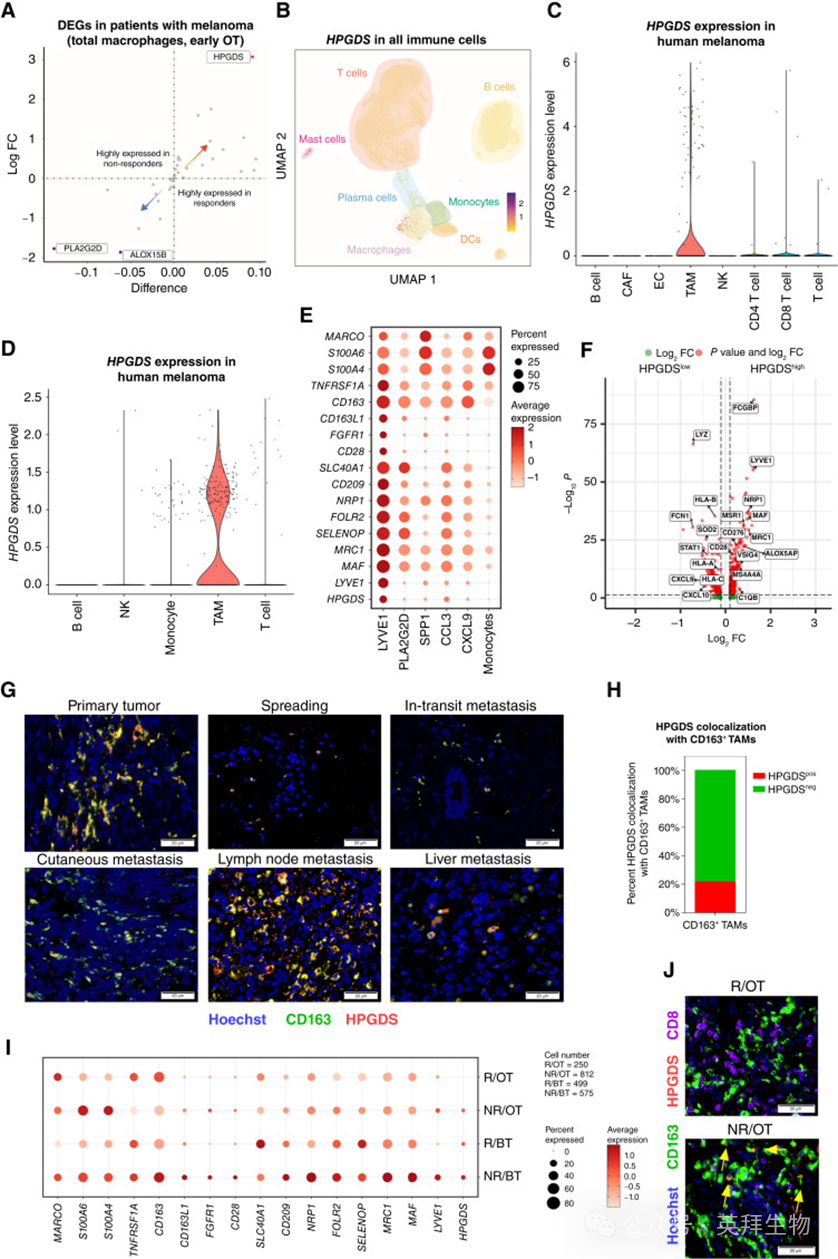
图1:在TAM子集中鉴定HPGDS
2 TAMs中的HPGDS促进免疫耐受和肿瘤前TME
对YUMM1.7小鼠黑色素瘤(携带临床相关突变Braf^V600E/+; Pten^−/−; Cdkn2^−/−;ref. 26)的公开scRNA-seq数据集分析显示,与人类情况类似,Hpgds主要在Mrc1_TAMs簇和少量肥大细胞簇中表达(附图 S2A)。在Ifn_TAMs(图2A)或单核细胞中未检测到Hpgds表达(图2B)。
为验证scRNA-seq数据,作者对YUMM 1.7黑色素瘤皮内移植模型的肿瘤微环境(TME)中不同细胞类型进行了分选。结果显示,Hpgds几乎仅在TAMs 中表达(附图 S2B)。与一项新的研究一致(ref. 27),Hpgds也可在淋巴结的滤泡辅助性T细胞中检测到,但其表达水平比 TAMs低约80倍(附图 S2C)。蛋白质水平验证表明,HPGDS 在 TAMs 中可检测到,而在其他肿瘤成分(如 CD8+ T 细胞、CD4+ T 细胞和 CD31+ 细胞)或健康器官(肺、肝)的组织巨噬细胞中均未检测到(附图 S2D、S2E)。在 TAMs 中,Hpgds 是仅有的 PGD2 合成酶,而 Lpgds 则选择性表达于血管内皮细胞(附图 S2F)。
支持“肿瘤来源的细胞因子诱导TAMs表达 HPGDS”的假说,作者观察到其表达在肿瘤条件培养基(TCM)中培养的骨髓来源巨噬细胞(BMDM)中显著上调(附图 S2G),在 IL-4、IL-6 或 IL-10 等促肿瘤细胞因子刺激下同样明显上调(图 2C)。然而,当去除促肿瘤细胞因子(如 IL-4)并加入促炎刺激(如 TNFα、LPS 或 LPS+IFNγ)后,Hpgds 表达被下调(图 2C);IFNγ 单独作用则无明显影响(图 2C)。一致地,ELISA 检测表明,IL-4 极化巨噬细胞培养上清液中 PGD2 浓度升高,而在给予 TNFα 24 小时后下降至对照水平(图 2D)。在 IL-4 极化的 BMDMs 中,与活化 T 细胞或其条件培养基共培养同样导致 Hpgds 下调,而中和 TNFα 则恢复其表达(图 2E;附图 S2H);相比之下,中和 IFNγ 抗体无此作用(附图 S2I)。
接着,作者在体内研究了 ICB 治疗反应性不同的黑色素瘤模型(ICB 抵抗性:YUMM 1.7;ICB 应答性:YUMMER 1.7;ref. 28)中,T 细胞耗竭或 αPD1 治疗对 TAMs 中 HPGDS 表达的调控作用(图 2F 和 G;附图 S2J–S2Q)。在抵抗性 YUMM 1.7 模型中,不论 CD8+ T 细胞耗竭还是 αPD1 治疗,肿瘤生长(附图 S2J–S2M)及 HPGDS+ 巨噬细胞数量均无变化(图 2H 和 I)。相反,在应答性 YUMMER 1.7 模型中,耗竭 CD8+ T 细胞(但不是 CD4+ T 细胞;附图 S3A–S3C)导致肿瘤进展(附图 S2N–S2Q)和 HPGDS+ 巨噬细胞积累(图 2H 和 I),而 αPD1 治疗则有效降低这两项指标(图 2H 和 I;附图 S2N、S2P)。无论是 αCD8 还是 αPD1,都未改变 TAM 总数(附图 S3D)。在应答性(但非抵抗性)模型中,αPD1 治疗导致 HPGDS+ TAMs 减少,这一效应依赖于 αPD1 对 CD8+ T 细胞的作用,因为在 αCD8 和 αPD1 联合处理时该效应消失(附图 S2N、S2Q)。与 Hpgds 与 Mrc1 共表达一致,HPGDS+ 巨噬细胞增加与 CD206+ TAMs 增加相关(图 2J)。此外,在 HPGDS+ TAMs 更为丰富的条件下,肿瘤血管密度更高且更异常(血管周长、尺寸和覆盖减少;图 2H–M);相反,当 HPGDS+ TAMs 较少时,肿瘤血管数量减少但呈“正常化”状态(图 2H–M;附图 S3E、S3F)。
随后,作者尝试通过药理学或遗传学方法阻止 HPGDS 的下调。首先,在 ICB 应答性 YUMMER 1.7 模型中联合使用 TNFα 拮抗剂 Enbrel 与 αPD1。结果表明,阻断 TNFα 使肿瘤对 αPD1 产生耐药性(图 2N;附图 S3G–S3I)。与单用 αPD1 不同,联合 Enbrel 的治疗使 HPGDS+ TAMs 密度恢复到对照水平,而 TAM 总浸润在各组间均无差异(图 2O–Q)。
基于此,作者进一步比较了不同遗传背景下免疫治疗的效果:对照组(Ctrl)与在 CD68 巨噬细胞特异性启动子驱动下持续过表达 Hpgds 的转基因小鼠(KI)(附图 S4A、S4B)。在 Ctrl 小鼠中,αPD1 有效抑制 YUMMER 1.7 肿瘤生长(图 3A、B;附图 S4C、S4D),并伴随 CD8+ T 细胞扩增(图 3C)和巨噬细胞中 Hpgds 的转录及蛋白水平降低(图 3D–F)。然而,在 KI 小鼠中,持续 Hpgds 表达导致明显的免疫治疗耐受(图 3A、B;附图 S4E、S4F),CD8+ T 细胞密度在 αPD1 治疗后无变化,甚至低于 Ctrl 小鼠 IgG 对照组(图 3C)。在 KI 小鼠的巨噬细胞中,HPGDS 水平升高且在 αPD1 治疗后不下降(图 3D–F),而 TAM 总数在各组间无差异(附图 S4G)。组织学检查显示,在 Ctrl 小鼠中,αPD1 应答与“更少但更优质”的血管相关(血管更大、覆盖和灌注更佳;图 3G–K)、肿瘤缺氧降低(图 3L、M)以及肺转移受抑制(图 3N);而在 KI 小鼠中,持续表达 Hpgds 足以诱导血管异常化和缺氧,即使在 αPD1 治疗下也如此(图 3L、M)。
这些数据提示,存在一个正反馈环路:活化 T 细胞释放 TNFα,下调免疫抑制性巨噬细胞中的 HPGDS,使其丧失对 T 细胞活化的抑制,从而促进免疫监视和抗肿瘤性 TME 的形成;相反,HPGDS 持续表达则塑造促肿瘤性 TME 并维持 αPD1 抵抗(图 3O)。
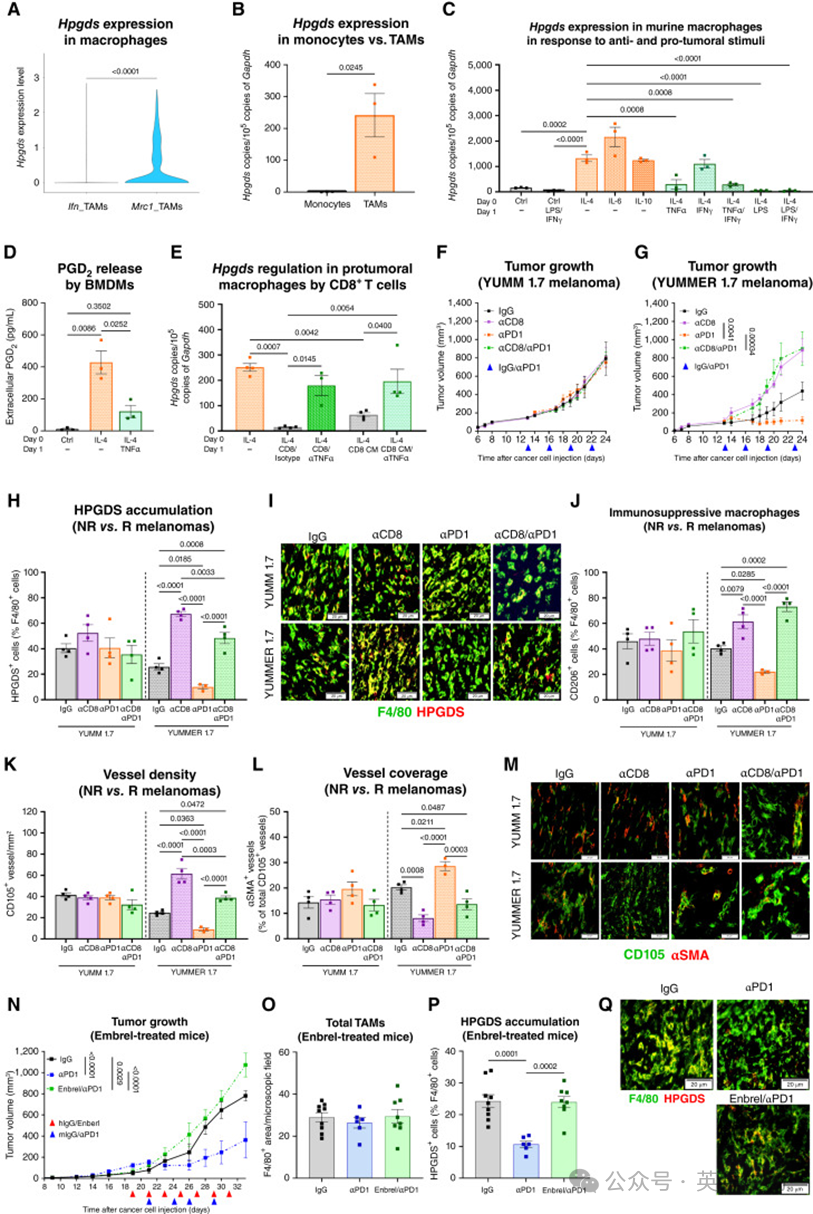
图2:HPGDS在巨噬细胞中的表达由原肿瘤细胞因子维持,并被活化的T细胞下调
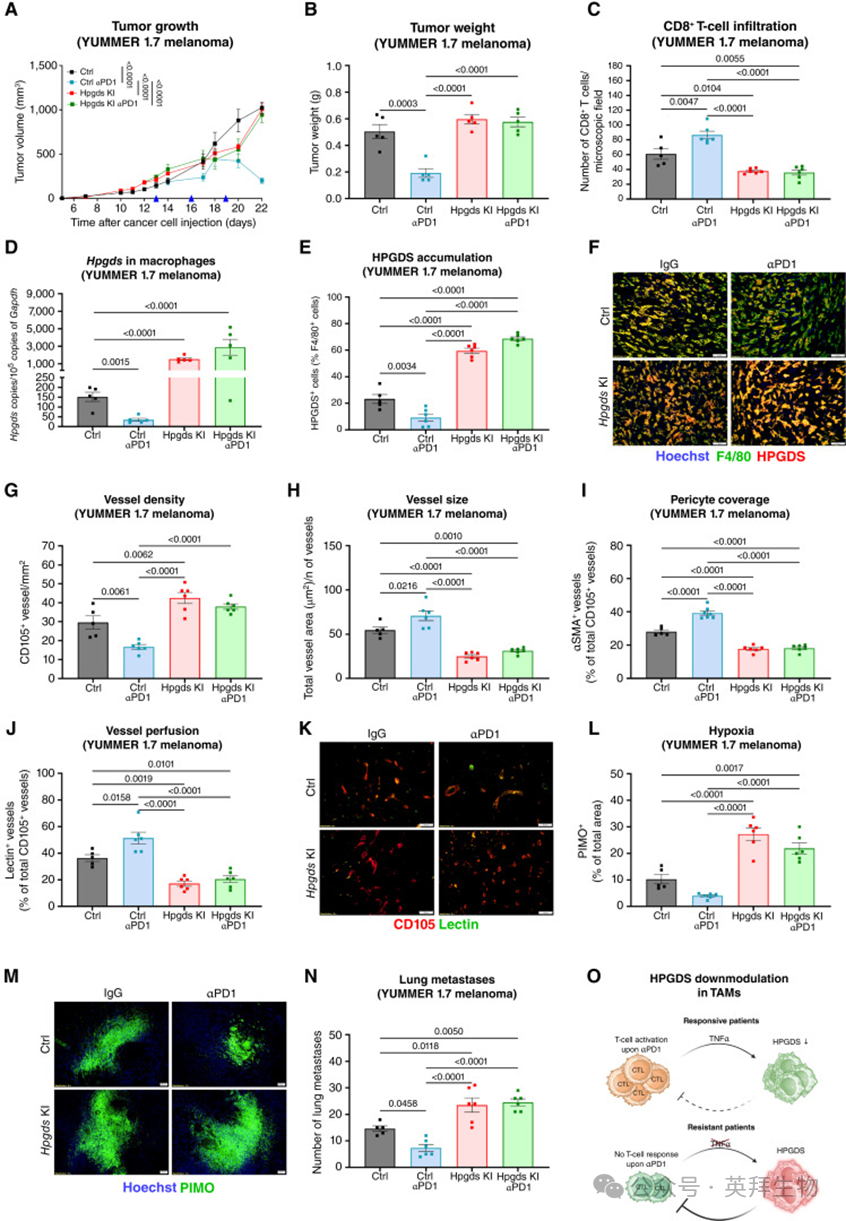
图3:巨噬细胞中Hpgds过表达可促进肿瘤进展并赋予ICB耐药性
3 TAMs中HPGDS的缺失塑造了TME的免疫监测、血管正常化和对免疫治疗的反应
作者构建了Hpgds条件性敲除小鼠,通过将 Hpgds floxed 小鼠与 CD64:Cre 系统杂交(CD64:Cre^Tg/W; Hpgds^L/L,简称 Hpgds^ΔMø),实现巨噬细胞特异性删除 Hpgds。作者在 YUMM 1.7 黑色素瘤皮内移植模型中分选 TAMs 并验证了删除效果(附图 S4H),同时在 Hpgds^ΔMø 与对照小鼠(CD64:Cre^W/W; Hpgds^L/L,简称 Ctrl)的骨髓来源巨噬细胞(BMDMs)中也得到了验证(附图 S4I)。利用质谱检测发现,在 Hpgds^ΔMø 小鼠的肿瘤间质液中,PGD2 水平显著下降,而 PGE2 未受影响(图 4A;附图 S4J);肿瘤裂解液及 BMDMs 中也观察到 PGD2 的类似下降(图 4B;附图 S4K)。
在监测YUMM 1.7 黑色素瘤的正位移植模型时,Hpgds^ΔMø 小鼠的肿瘤体积和重量分别比对照鼠降低约 80% 和 60%,其中 9 只小鼠中有 4 只表现出肿瘤完全排斥(图 4C、D;附图 S4L、S4M)。此外,循环肿瘤细胞和肺部转移灶在 Hpgds^ΔMø 小鼠中显著减少(图 4E–G;附图 S4N)。
对TAMs的整体转录组无偏分析显示,Hpgds 敲除后,其免疫抑制和血管生成特征均减弱(附图 S4O)。作者通过 qRT-PCR 对部分差异基因进行了独立验证,如 Cxcl9、Cxcl10、Cxcl11、Cd80、Fgf2 和 Hgf(附图 S4P–S4U)。流式细胞术和组织学分析进一步确认:Hpgds^ΔMø 小鼠肿瘤中的 CD206+、CD204+ 和 MHC-II^low 巨噬细胞减少,而 CD11c、CD80、CD86 和 MHC-II 高表达巨噬细胞增加(图 4H–J),但 TAM 总数未见变化(附图 S5A、S5B)。此外,CD8+ T 细胞浸润增加,并伴随更高的激活/效应标志物(CD69、IFNγ、GZMB)(图 4K、L);相比之下,常规树突状细胞(cDC)、中性粒细胞和 CD4+ T 细胞未受影响(附图 S5C–S5G)。在血管结构方面,Hpgds^ΔMø 小鼠肿瘤的血管数量减少,但更大,且伴随周细胞覆盖和灌注改善(图 4M–U)。
为明确CD8+ T 细胞在肿瘤抑制及转移阻止中的作用,作者给予Hpgds^ΔMø 小鼠 CD8 耗竭抗体(附图 S5H)。结果显示,CD8 耗竭使 Hpgds^ΔMø 小鼠的肿瘤生长恢复至对照水平(图 4V、W;附图 S5I–S5L)。然而,CD8 耗竭并未影响 CD80+ TAMs 的比例、血管正常化或肺部转移抑制(图 4X、Y;附图 S5M–S5O)。因此,CD8+ T 细胞是 Hpgds^ΔMø 小鼠中肿瘤生长抑制所必需的,而血管正常化和转移抑制则归因于巨噬细胞的重编程。最后,作者在 Ctrl 和 Hpgds^ΔMø 小鼠中皮内接种 YUMM 1.7 黑色素瘤细胞,并在肿瘤平均体积达到 150 mm³ 时给予αPD1治疗(图 5A、B)。结果显示,Ctrl小鼠对αPD1治疗耐药,而 Hpgds删除可使肿瘤对 αPD1 敏感并防止复发(图 5A、B;附图 S5P–S5S)。
为进一步验证这一发现,作者利用可诱导的巨噬细胞特异性 CRISPR/Cas9 系统改造供体小鼠造血祖细胞,建立了完全重建的 WT→WT 与 Hpgds 敲除→WT 嵌合体(附图 S6A、S6B)。在这些小鼠中皮内接种 YUMM 1.7 黑色素瘤细胞,结果确认了 TAMs 中 Hpgds 的蛋白水平敲除(组织学与 WB,附图 S6C、S6D)。Hpgds 敲除显著抑制了肿瘤的生长(附图 S6E–S6G)和重量(附图 S6H、S6I),并导致 TAMs 从促肿瘤向抗肿瘤表型转变(附图 S6K–S6N),同时 CD8+ T 细胞数量增加(附图 S6O、S6P),这些细胞重新定位至肿瘤核心区域并表现出更高的 IFNγ 和 GZMB 表达(附图 S6Q–S6S)。综上,这些结果表明:在 TAMs 中抑制 HPGDS 能够重塑TME,从而阻止肿瘤生长和转移,并使肿瘤对αPD1 敏感。
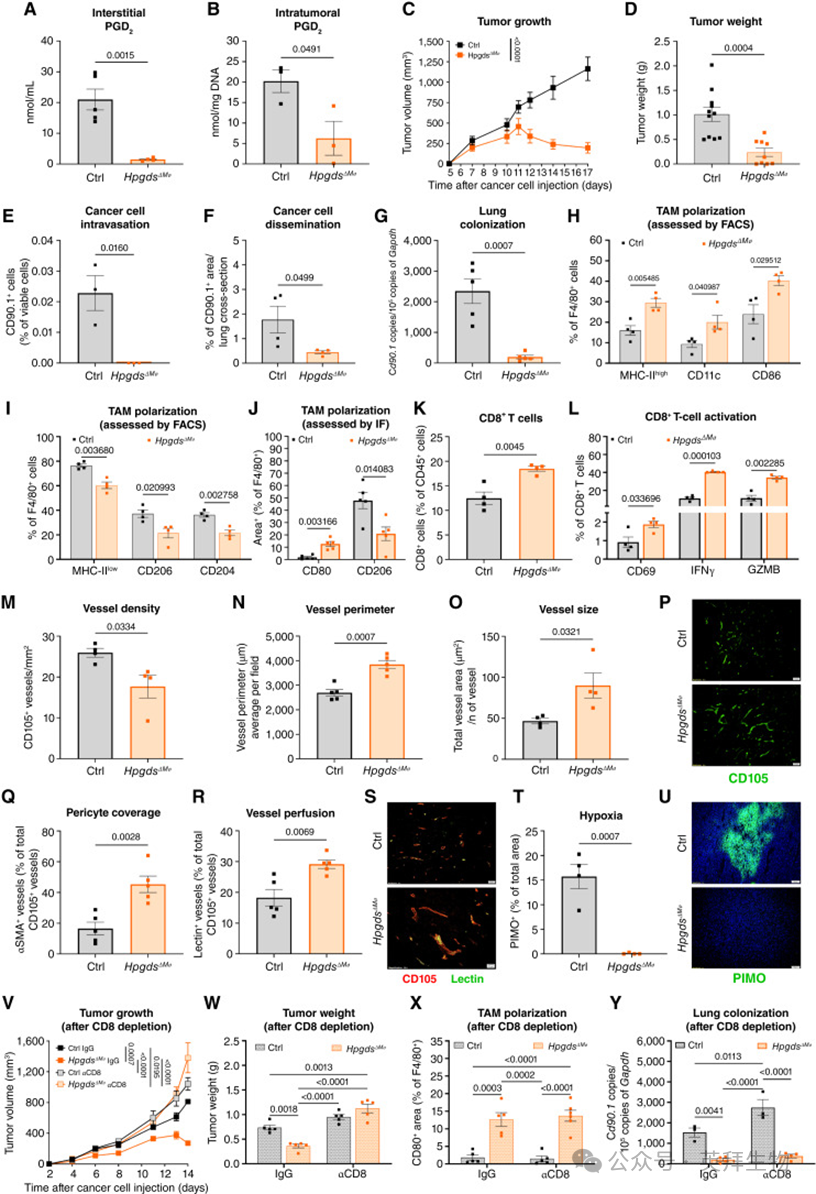
图4:TAMs中的Hpgds缺失重塑TME并抑制肿瘤生长
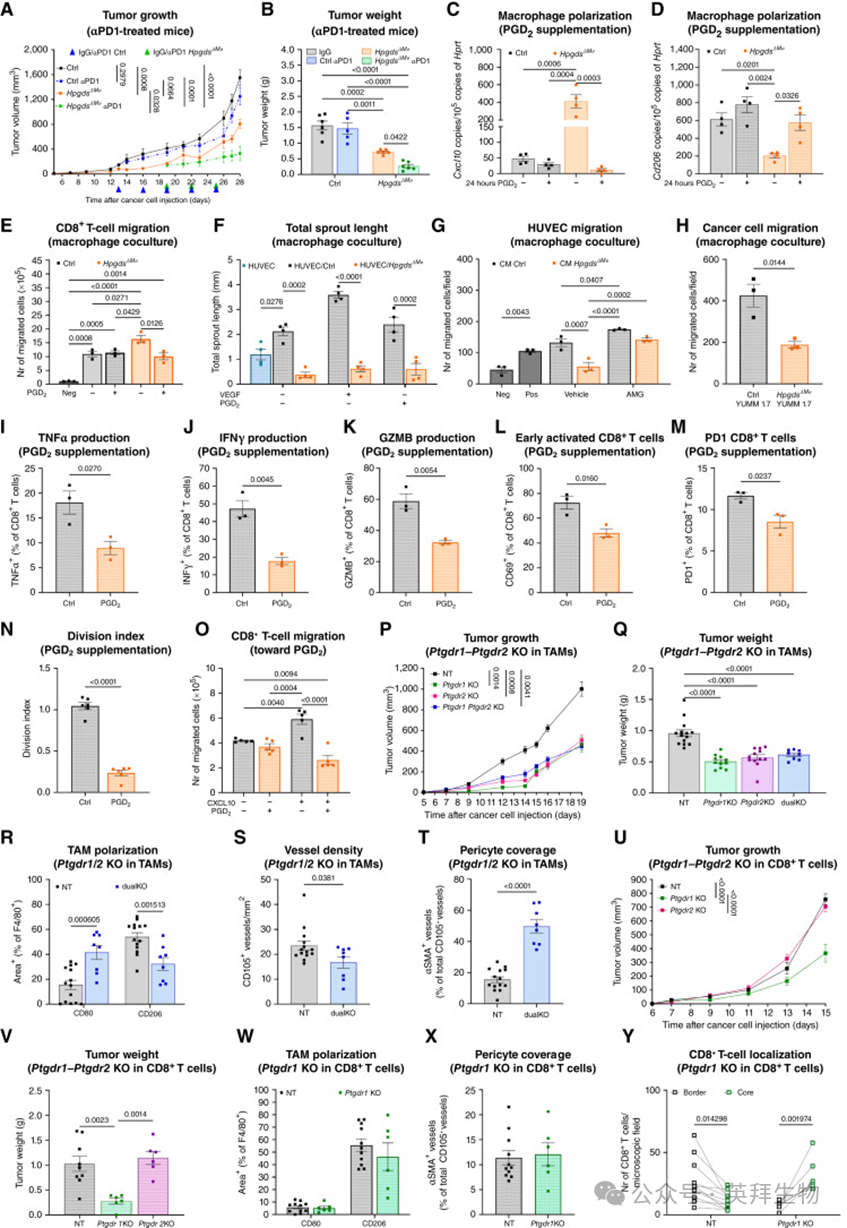
图5:Hpgds缺失与ICB的协同作用以及PGD2信号传导对TAM再教育和 CD8+ T细胞生物学的贡献
4 HPGDS/PGD2信号抑制的直接和间接作用
PGD2的下游效应通过激活两种G蛋白偶联受体来介导,分别是 DP1(由 PTGDR1 编码)和DP2(由 PTGDR2 编码)。在YUMM 1.7 黑色素瘤肿瘤中对这些受体的表达分析显示,在检测的细胞类型中,Ptgdr1 和 Ptgdr2 在 CD8+ T 细胞、CD4+ T 细胞、巨噬细胞和中性粒细胞中的表达较高,而在内皮细胞(ECs)和肿瘤细胞中则低或不可检测(附图 S7A、S7B)。因此,作者重点分析了 PGD2 对 CD8+ T 细胞和巨噬细胞的作用,而未进一步研究 CD4+ T 细胞和中性粒细胞,因为在 Hpgds^ΔMø 小鼠中它们未表现出变化。
已有研究表明,PGD2 拮抗剂可赋予巨噬细胞促炎表型(ref. 36)。基于作者在 TAMs 中的发现,以及在 Hpgds^ΔMø 小鼠中 CD8+ T 细胞耗竭后肿瘤血管仍然保持正常化的现象,作者推测:由 HPGDS+ TAMs 分泌的 PGD2 可能通过影响巨噬细胞行为,间接导致血管异常化。一致地,在体外实验中,IL-4 极化的 Hpgds^ΔMø BMDMs 表现出更低的抗炎/促血管因子水平和更高的促炎/血管稳定因子水平(附图 S7C)。通过 qRT-PCR 进一步验证,PGD2 补充可下调 BMDMs 中 Cxcl10 表达并上调 Mrc1/CD206(图 5C、D),但对 Hpgds 自身的表达无显著影响(附图 S7D)。在功能检测中,CD8+ T 细胞更倾向迁移至 Hpgds^ΔMø BMDMs,而 PGD2 预处理的巨噬细胞则削弱了这一效应(图 5E)。
为研究Hpgds^ΔMø BMDMs 在血管正常化中的功能意义,作者在 I 型胶原凝胶中开展了3D血管芽生实验。结果显示,与内皮细胞共培养时,Hpgds^ΔMø BMDMs 可显著减少血管芽的数量和长度(图 5F;附图 S7E)。此外,BMDMs 条件培养基对 ECs 迁移的调控作用也减弱,表明其呈现更强的抗血管生成表型;添加 FGF2 或 HGF 能部分恢复 ECs 的迁移能力(附图 S7F)。类似地,当将 HUVECs 预处理于含有 CXCR3 抑制剂的 BMDM 条件培养基中,再给予 VEGF 刺激时,也能恢复 ECs 的迁移(图 5G;附图 S7G)。这些结果提示,Hpgds 缺失的巨噬细胞通过负向/正向调控关键因子,从而影响内皮细胞行为。此外,YUMM 1.7 黑色素瘤细胞向 Hpgds^ΔMø BMDMs 的迁移也受损(图 5H)。值得注意的是,PGD2 对 ECs 球体或 YUMM 1.7 黑色素瘤细胞本身的芽生或迁移无直接影响(附图 S7H–S7K)。这些数据提示,HPGDS 介导的 PGD2 主要通过自分泌方式调节巨噬细胞极化,但不能排除 PGD2 对 CD8+ T 细胞的直接作用。
为探究 PGD2对CD8+ T 细胞的直接效应,作者在预活化的 CD8+ T 细胞培养基中补充 PGD2 并用 PMA/离子霉素再刺激。结果显示:TNFα、IFNγ 和 GZMB 的分泌减少(图 5I–K),同时 CD69 和 PD1 的表达水平下降(图 5L、M)。在功能上,OVA 特异性的OT-I T 细胞在 PGD2 处理后,其增殖能力和对表达 OVA 的 YUMM 1.7 细胞的杀伤作用均受损(图 5N;附图 S7L、S7M)。与此一致,PGD2 可显著抑制活化 T 细胞的 CXCL10 诱导迁移(图 5O)。因此,PGD2 既可通过直接作用于 CD8+ T 细胞,又可通过巨噬细胞介导的间接机制发挥免疫抑制效应。
考虑到PGD2通过DP1和DP2 发挥作用,作者进一步在体内评估这两种受体在 TAM 重编程中的作用。利用 CRISPR/Cas9 技术,作者在巨噬细胞中分别敲低 Ptgdr1 或 Ptgdr2(附图 S8A),并通过 BMDMs 验证了转录水平的敲降效果(附图 S8B)。在免疫重建小鼠体内接种 YUMM 1.7 黑色素瘤细胞后发现,无论靶向 Ptgdr1 还是 Ptgdr2,都可显著降低肿瘤生长和重量(图 5P、Q)。两者联合靶向并未产生叠加效应(图 5P、Q;附图 S8C–S8F),提示单独阻断 DP1 或 DP2 就足以发挥治疗作用,可能因这两个受体间存在分子互作(ref. 37)。在这些肿瘤中,虽然 TAM 总数未变(附图 S8G),但其 CD80 表达上调而 CD206 下调(图 5R),同时肿瘤血管数量更少且被周细胞覆盖更好(图 5S、T)。然而,T 细胞在肿瘤核心的募集未受影响(附图 S8H)。相应地,在 Hpgds^ΔMø 小鼠中再次靶向 Ptgdr1 或 Ptgdr2 并未产生额外效应(附图 S8I–S8M)。综上,这些结果表明:PGD2 对 HPGDS+ 巨噬细胞的自分泌作用依赖 DP1 和 DP2 的共同参与,从而维持其免疫抑制表型。
5 药理学HPGDS抑制重塑TME并阻断肿瘤生长
为探索HPGDS的治疗靶向潜能,作者测试了HPGDS 抑制剂 HQL-79,该抑制剂已在临床前研究中被证实可降低肿瘤转移(refs. 38, 39)。在 YUMM 1.7黑色素瘤小鼠模型中,单独使用 HQL-79 并不能显著影响肿瘤生长(图 6A,左)。然而,当与αPD1联合使用时,HQL-79与αPD1 协同抑制肿瘤生长,部分小鼠甚至出现完全应答(图 6A,右)。在联合治疗组中,肿瘤重量降低(图 6B)、CD8+ T 细胞数量和功能增强(图 6C、D),而TAM总数未变(附图 S9A)。
作者还研究了DP1拮抗剂laropiprant与αPD1 的联合应用。类似地,laropiprant 单独使用无明显效果(图 6E,左),但与 αPD1 联合时显著抑制肿瘤生长(图 6E,右),并导致肿瘤重量下降(图 6F)和 CD8+ T 细胞浸润增加(附图 S9B)。组织学分析进一步表明,联合治疗组肿瘤中 HPGDS+ TAMs 显著减少,而 CD8+ T 细胞和 CD80+ TAMs 增加(图 6G–I),并且肿瘤血管减少但更大且被周细胞覆盖(图 6J、K;附图 S9C)。
为评估PGD2阻断的临床相关性,作者在人黑色素瘤类器官模型中测试了 laropiprant。结果显示:在来自 ICB 无应答患者的类器官培养物中,laropiprant 与αPD1联合可增强 CD8+ T 细胞的扩增和活化(CD69 表达增加)(图 6L、M),并促进 GZMB+ T 细胞的募集和浸润(图 6N、O)。相反,在来自 ICB 应答患者的类器官中,laropiprant 并未进一步增强 αPD1 的作用(附图 S9D–S9H)。
综上所述,这些结果表明:药理学抑制 HPGDS/PGD2 信号通路可作为一种可行策略,增强αPD1的疗效,尤其适用于免疫检查点治疗无应答的黑色素瘤患者。
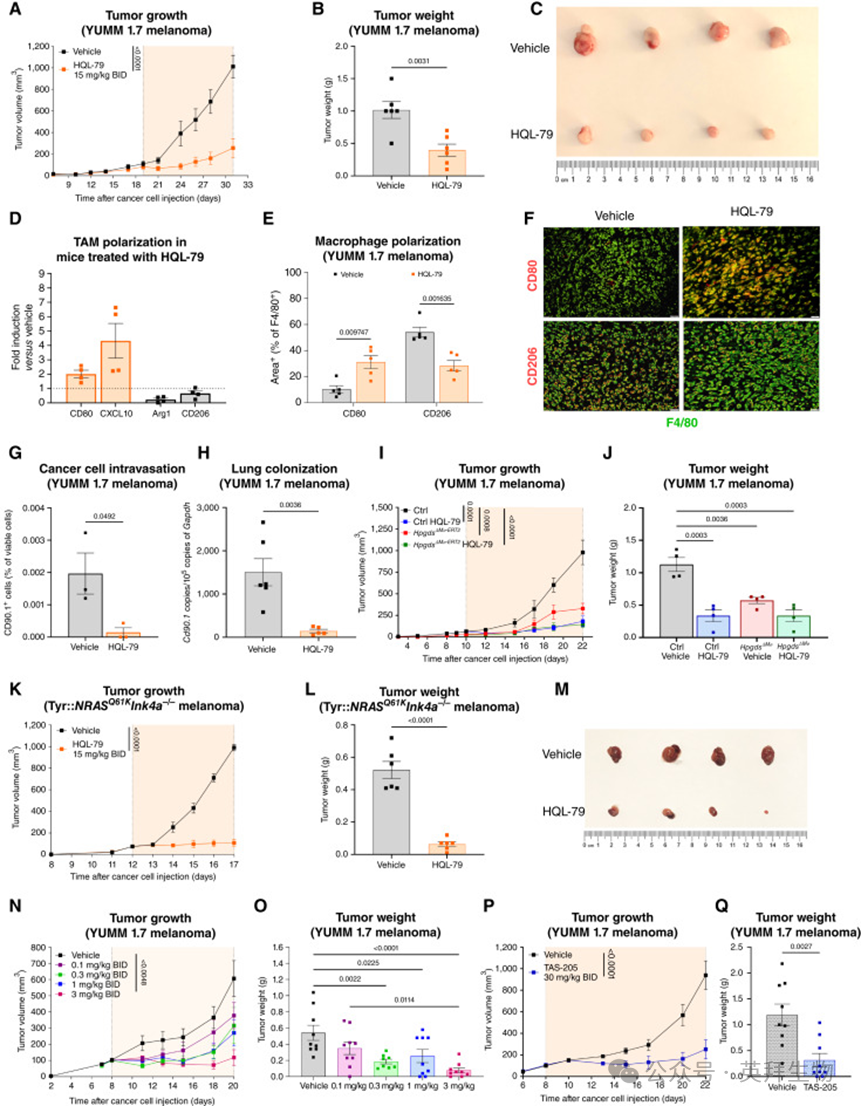
图6:药理学HPGDS抑制表型复制其基因缺失在TAM中的影响。
6 在临床相关小鼠模型和人类环境中进一步验证HPGDS的药理学抑制作用
为探究HPGDS+ TAMs 是否仅限于黑色素瘤,作者分析了一个整合的 scRNA-seq 数据集,该数据集包含 11 种不同癌症类型(共 221 例患者,24 种癌症亚型)的 30 万多个细胞(ref. 40)。结果显示,HPGDS 在多种癌症类型中的 TAMs 中均有表达,但在其他免疫细胞亚群中几乎不表达(图 7A;附图 S10A)。具体而言,在肺癌、结直肠癌、肾癌、胃癌和肝癌中均可检测到 HPGDS+ TAMs(图 7B)。
随后,作者利用 The Cancer Genome Atlas (TCGA) 数据集分析了 HPGDS 的临床相关性。结果发现,HPGDS 表达水平与多种癌症类型的不良预后密切相关,包括黑色素瘤、肺癌、肝癌和胃癌(图 7C)。一致地,在作者收集的人类黑色素瘤、肺癌、肝癌和胃癌组织的免疫组化(IHC)分析中,约 15%–25% 的 CD163+ TAMs 与 HPGDS 共定位(图 7D;附图 S10B)。进一步分析表明,在这些癌症类型中,HPGDS+ TAMs 的浸润与 CD8+ T 细胞缺乏相关,并伴随血管异常化和免疫抑制性微环境(图 7E–G;附图 S10C–S10F)。因此,这些结果表明:HPGDS/PGD2 驱动的免疫耐受并非黑色素瘤特有,而是多种实体瘤中的一种共同机制。
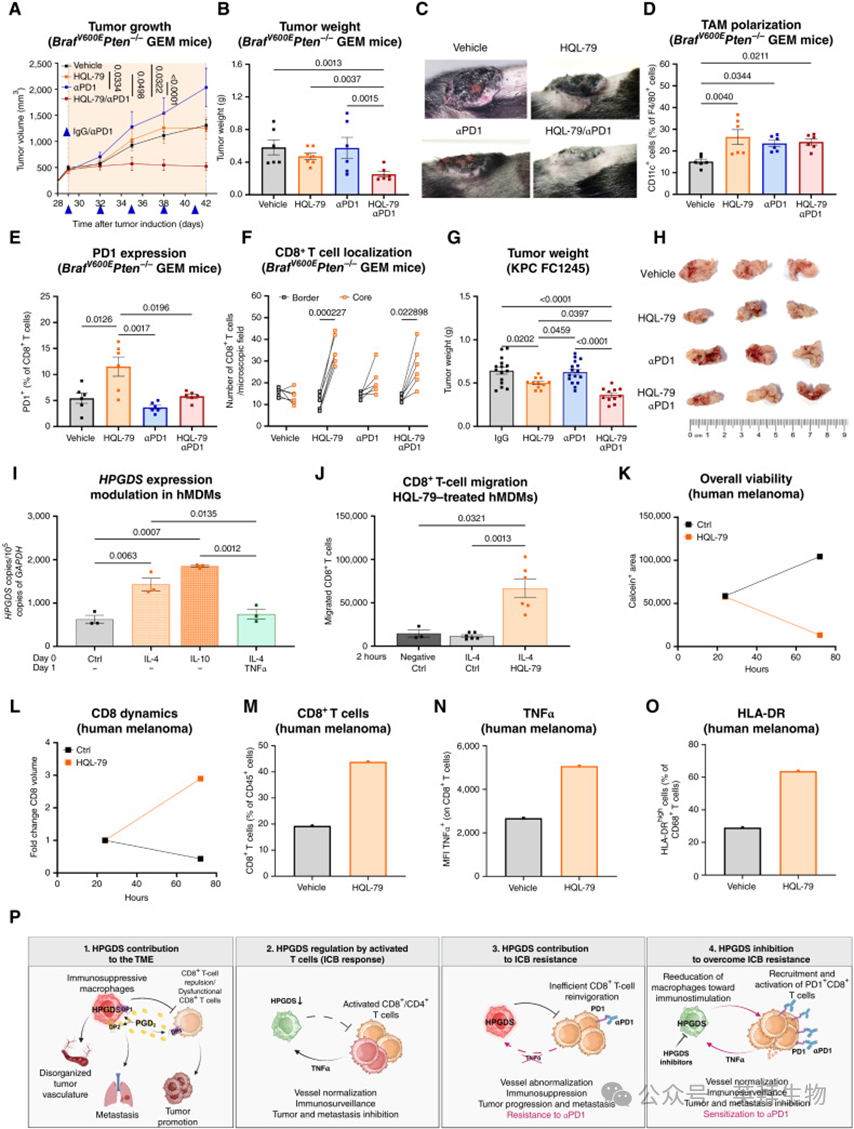
图7:HPGDS抑制克服免疫治疗的耐药性,并且在人类环境中有效
结论:
总之,作者表明活化的T细胞下调TAMs中的HPGDS可以控制先天免疫系统,并通过减少PGD2的产生来塑造整个TME。一致地,作者提供的证据表明,靶向黑色素瘤中的HPGDS-PGD2-DP1/DP2轴可以使TAM远离其免疫抑制和肿瘤前行为,促进免疫许可和抗肿瘤TME。
参考文献:
Trotta R, Rivis S, Zhao S, Orban MP, Trusso Cafarello S, Charatsidou I, Pozniak J, Dehairs J, Vanheer L, Pulido Vicuna CA, Boecxstaens V, Bechter O, Bosisio FM, Swinnen JV, Marine JC, Mazzone M. Activated T Cells Break Tumor Immunosuppression by Macrophage Reeducation. Cancer Discov. 2025 Jul 3;15(7):1410-1436. doi: 10.1158/2159-8290.CD-24-0415. PMID: 40094380; PMCID: PMC12223510.