






Immunity:XBP1缺失破坏黏液屏障,协同坏死性凋亡恶化结肠炎
内质网(ER)应激与坏死性凋亡(necroptosis)在炎症性肠病(IBD)的发病机制中具有关联性,但这些通路之间的潜在交互作用尚不明确。本研究显示,在肠道上皮细胞(IEC)特异性缺失caspase-8或其衔接蛋白FADD(Fas associated with death domain)的小鼠中,X盒结合蛋白1(XBP1)的缺失会显著加剧坏死性凋亡诱导的结肠炎(而非回肠炎)的发展。从机制上讲,XBP1缺失导致结肠中黏蛋白2(MUC2)表达减少和黏液层形成受损,使得细菌能够穿透并抵达上皮表面。在上皮单层结构完整的情况下,这并不足以引发结肠炎,但与IEC坏死性凋亡协同作用后会诱发严重结肠炎症。我们的研究结果揭示,XBP1和caspase-8分别调控肠道屏障的不同组成部分,通过协同作用维持黏膜免疫稳态并预防结肠炎症。这一发现有助于更好地理解IBD的致病机制。本文于2025年8月发表于《Immunity》,IF 26.3。
图形摘要
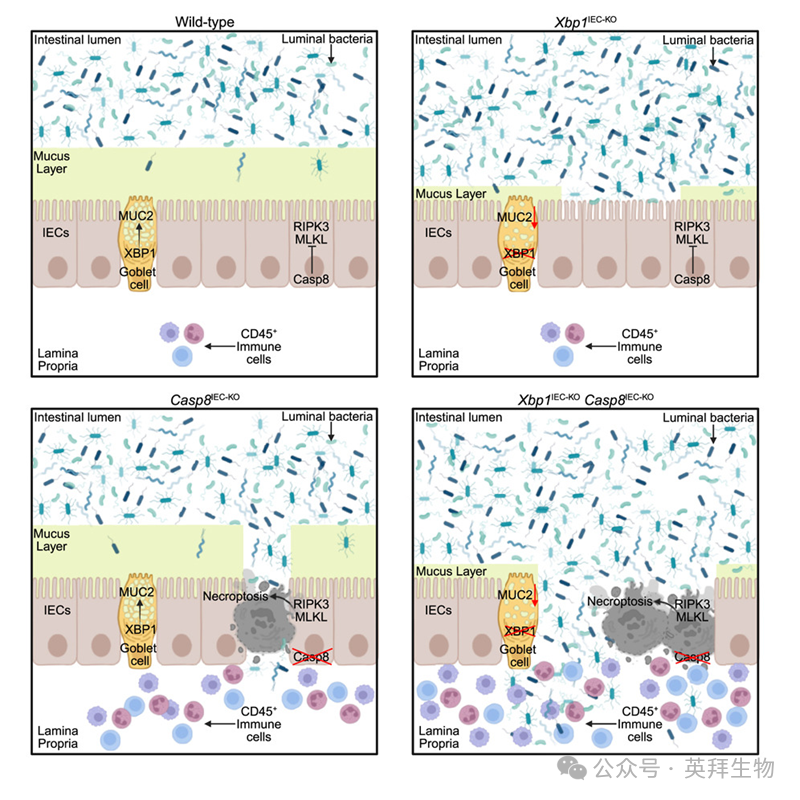
主要研究结果
1.XBP1抑制Casp8IEC-KO和FaddIEC-KO小鼠的坏死性凋亡诱导性结肠炎发展
为研究XBP1和caspase-8在IECs中的作用,我们构建了Xbp1fl/fl × Vil1-Cre(Xbp1IEC-KO)和Casp8fl/fl × Vil1-Cre(Casp8IEC-KO)小鼠。与既往报道一致,Xbp1IEC-KO小鼠结肠未出现自发病理改变,而Casp8IEC-KO小鼠表现出轻度至重度结肠炎,特征包括增生、轻度上皮侵蚀、IEC死亡及免疫细胞浸润(图1A)。为评估XBP1与caspase-8在调节肠上皮稳态中的潜在交互作用,我们构建并分析了Xbp1IEC-KOCasp8IEC-KO小鼠(IECs中特异性缺失XBP1和caspase-8)。与Casp8IEC-KO及Xbp1fl/fl × Casp8fl/WT × Villin-cretg/WT(Xbp1IEC-KOCasp8IEC-Het)小鼠相比,Xbp1IEC-KOCasp8IEC-KO小鼠体重显著减轻(图1B),表明IECs中XBP1与caspase-8的联合缺失导致严重肠道病理改变。组织学分析显示,Xbp1IEC-KOCasp8IEC-KO小鼠出现严重结肠炎,表现为上皮过度增殖、IEC死亡数量增加、广泛上皮侵蚀形成全结肠散布的溃疡、黏膜及黏膜下层免疫细胞浸润增加,以及大量隐窝脓肿形成(图1A)。组织学结肠炎评分、溃疡形成及上皮细胞增殖的量化分析证实,与Casp8IEC-KO小鼠相比,Xbp1IEC-KOCasp8IEC-KO小鼠结肠病理恶化程度更高(图1C–1E)。RNA测序(RNA-seq)显示,与Xbp1IEC-KO和Casp8IEC-KO小鼠相比,Xbp1IEC-KOCasp8IEC-KO小鼠结肠炎症基因表达上调,进一步支持双基因缺失比单基因缺失导致更严重的结肠炎症(图1F)。我们前期研究表明,通过RIPK3或MLKL基因缺失抑制坏死性凋亡可阻止Casp8IEC-KO小鼠结肠炎发展。因此我们探讨:在XBP1介导的未折叠蛋白反应(UPR)与caspase-8信号联合抑制后,坏死性凋亡是否参与加剧结肠炎。为此,我们构建并分析了Xbp1IEC-KOCasp8IEC-KORipk3−/−和Xbp1IEC-KOCasp8IEC-KOMlkl−/−小鼠。大体观察、免疫组织学及RNA-seq分析表明,抑制RIPK3-MLKL介导的坏死性凋亡可完全恢复Xbp1IEC-KOCasp8IEC-KO小鼠体重并阻止结肠炎发展(图1A和1C–1G)。IEC特异性缺失FADD的小鼠会发生自发性结肠炎,且严重程度高于Casp8IEC-KO小鼠。与Xbp1IEC-KOCasp8IEC-KO小鼠的研究结果一致,并行免疫组织学评估与量化分析表明,Xbp1IEC-KOFaddIEC-KO小鼠比FaddIEC-KO小鼠发生更严重的结肠炎。RIPK3缺失同样阻止了Xbp1IEC-KOFaddIEC-KORipk3−/−小鼠结肠炎的发展。综上所述,这些结果表明在上皮细胞中,XBP1通过抑制坏死性凋亡诱导的炎症性结肠炎,在FaddIEC-KO和Casp8IEC-KO小鼠中发挥保护作用。
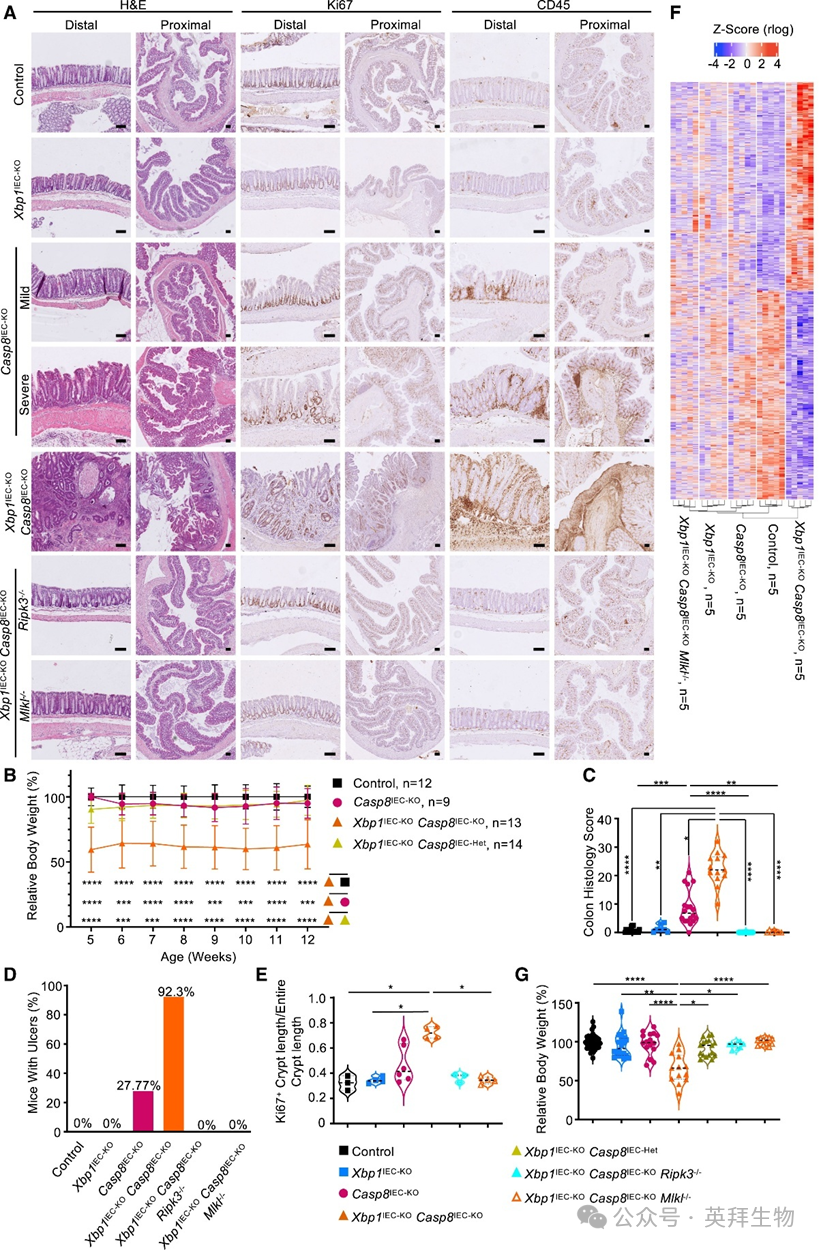
图1 IEC特异性XBP1缺失加剧Casp8IEC-KO小鼠的坏死性凋亡诱导性结肠炎
2.XBP1缺失不会加剧Casp8IEC-KO和FaddIEC-KO小鼠的回肠炎
Xbp1IEC-KO小鼠会发生自发性回肠炎,表现为上皮增生、潘氏细胞缺失和黏膜免疫细胞浸润。此外,Casp8IEC-KO小鼠会发生依赖RIPK3-MLKL介导的坏死性凋亡的自发性回肠炎。为评估XBP1与caspase-8联合缺失是否也会加剧回肠炎,我们比较了Xbp1IEC-KO、Casp8IEC-KO和Xbp1IEC-KOCasp8IEC-KO小鼠的回肠病理。对Xbp1IEC-KO小鼠回肠切片的溶菌酶免疫染色显示隐窝中潘氏细胞数量减少,这与潘氏细胞特异性基因(包括溶菌酶Lyz1、Defa5和Defa-rs10)mRNA表达降低相关(图2A和2B)。但我们在Xbp1IEC-KO小鼠回肠中未观察到CD45免疫细胞浸润显著增加(这与既往研究报道不同),提示遗传背景和/或微生物群的差异可能影响这些动物的小肠病理(图2A)。与既往报道一致,Casp8IEC-KO小鼠出现自发性回肠炎,表现为潘氏细胞几乎完全缺失、上皮增生、IEC死亡及免疫细胞浸润增加(图2A和2B)。Xbp1IEC-KOCasp8IEC-KO小鼠也发生自发性回肠炎,特征包括上皮增生、潘氏细胞缺失、IEC死亡及固有层免疫细胞浸润(图2A–2D)。并行组织病理学和基因表达分析表明,Xbp1IEC-KOCasp8IEC-KO小鼠的小肠病理与Casp8IEC-KO小鼠的回肠炎高度相似(图2A–2D)。尽管Xbp1IEC-KO小鼠回肠组织学评分显著升高(图2C),但这主要归因于该小鼠模型相关的隐窝增生(图2D)。此外,Xbp1IEC-KOFaddIEC-KO小鼠的小肠病理也与FaddIEC-KO小鼠的回肠炎高度相似。这些结果表明,与结肠中XBP1缺失会加剧坏死性凋亡诱导的炎症形成鲜明对比,IECs中XBP1缺失并未改变Casp8IEC-KO和FaddIEC-KO小鼠回肠炎的严重程度。
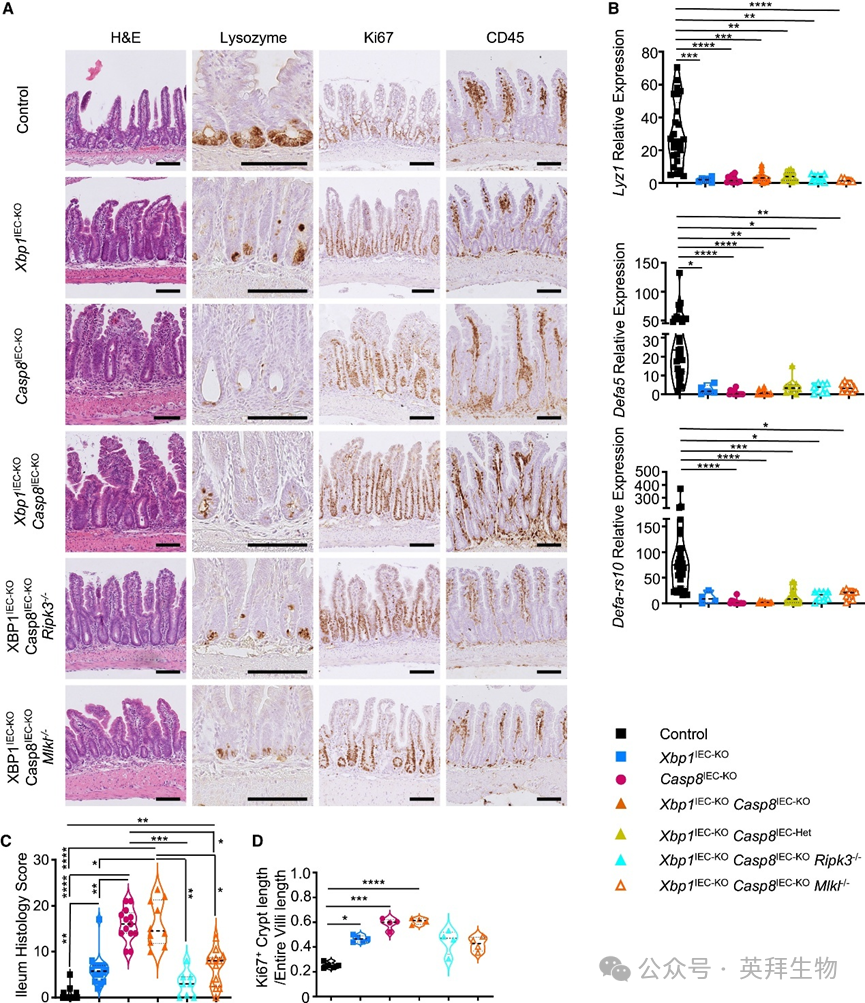
图2 抑制caspase-8介导的凋亡和RIPK3-MLKL诱导的坏死性凋亡不能阻止XBP1缺失所致的潘氏细胞丢失
3.联合抑制凋亡和坏死性凋亡不能阻止Xbp1IEC-KO小鼠的潘氏细胞丢失
有观点认为IEC凋亡参与Xbp1IEC-KO小鼠回肠的病理发展。为探究caspase-8介导的凋亡和RIPK3-MLKL诱导的坏死性凋亡是否导致Xbp1IEC-KO小鼠的上皮增生和潘氏细胞丢失,我们检测了Xbp1IEC-KOCasp8IEC-KORipk3−/−、Xbp1IEC-KOCasp8IEC-KOMlkl−/−和Xbp1IEC-KOFaddIEC-KORipk3−/−动物的小肠组织。免疫组织学与基因表达分析显示,RIPK3或MLKL缺失将Xbp1IEC-KOCasp8IEC-KO和Xbp1IEC-KOFaddIEC-KO小鼠回肠炎严重程度降低至Xbp1IEC-KO小鼠水平(图2A–2D)。但Ki67和溶菌酶免疫染色表明,联合抑制caspase-8依赖性凋亡和RIPK3-MLKL依赖性坏死性凋亡并不能阻止XBP1缺失诱导的上皮增生和潘氏细胞丢失——Xbp1IEC-KOCasp8IEC-KORipk3−/−、Xbp1IEC-KOCasp8IEC-KOMlkl−/−和Xbp1IEC-KOFaddIEC-KORipk3−/−小鼠均表现出与Xbp1IEC-KO小鼠相似的小肠病理(图2A–2D)。与溶菌酶免疫染色结果一致,定量RT-PCR(RT-qPCR)分析显示Xbp1IEC-KOCasp8IEC-KORipk3−/−和Xbp1IEC-KOCasp8IEC-KOMlkl−/−小鼠中Lyz1、Defa5和Defa-rs10表达显著降低(图2B)。我们推测不依赖caspase-8和坏死性凋亡的细胞死亡可能参与XBP1缺失诱导的潘氏细胞丢失和上皮增生。因此我们对Xbp1IEC-KOCasp8IEC-KORipk3−/−、Xbp1IEC-KOCasp8IEC-KOMlkl−/−和Xbp1IEC-KOFaddIEC-KORipk3−/−小鼠回肠切片进行cleaved caspase-3(CC3)和TUNEL染色。这些实验未发现Xbp1IEC-KO、Xbp1IEC-KOCasp8IEC-KORipk3−/−和Xbp1IEC-KOCasp8IEC-KOMlkl−/−小鼠隐窝内死亡IECs数量较对照组增加。与既往发现一致,我们检测到Xbp1IEC-KOFaddIEC-KORipk3−/−小鼠回肠中CC3和TUNEL阳性IECs数量较对照组增加。这些结果表明FADD-caspase-8依赖性凋亡和RIPK3-MLKL介导的坏死性凋亡不驱动Xbp1IEC-KO小鼠的潘氏细胞丢失和上皮增生。
4.上皮细胞固有的TNFR1信号驱动Xbp1IEC-KOCasp8IEC-KO小鼠的结肠炎恶化
IEC特异性肿瘤坏死因子受体1(TNFR1)缺失可减轻Casp8IEC-KO小鼠的结肠炎发展。TNFR1也参与Xbp1IEC-KO小鼠回肠炎的发展。为探究IECs中TNFR1信号是否介导Xbp1IEC-KOCasp8IEC-KO小鼠的严重结肠炎,我们构建并分析了Xbp1IEC-KOCasp8IEC-KOTnfr1IEC-KO小鼠。免疫组织学评估和RNA-seq分析显示,上皮TNFR1缺失强烈抑制了Xbp1IEC-KOCasp8IEC-KOTnfr1IEC-KO小鼠的结肠炎发展(图3A–3E)。有观点认为,XBP1缺失的IECs和/或潘氏细胞中核因子κB(NF-κB)激活可能驱动肿瘤坏死因子(TNF)产生,从而参与Xbp1IEC-KO小鼠的肠炎发生。因此我们推测IEC来源的TNF水平升高可能加剧Xbp1IEC-KOCasp8IEC-KO小鼠的结肠炎。为验证该假设,我们构建并分析了IECs中特异性缺失XBP1、caspase-8和TNF的Xbp1IEC-KOCasp8IEC-KOTnfIEC-KO小鼠。结肠切片免疫组织学分析表明,Xbp1IEC-KOCasp8IEC-KOTnfIEC-KO小鼠发生的结肠炎严重程度与Xbp1IEC-KOCasp8IEC-KO小鼠相似(图3A–3D),排除了IEC来源的TNF在驱动该病理中的作用。此外,IEC特异性缺失TNF或TNFR1并未缓解Xbp1IEC-KOCasp8IEC-KO小鼠的潘氏细胞丢失和小肠病理。这些结果与我们之前的发现一致:TNFR1在Casp8IEC-KO小鼠坏死性凋亡驱动的小肠炎症中不起重要作用。综上结果表明,上皮细胞固有的TNFR1信号驱动Xbp1IEC-KOCasp8IEC-KO小鼠的结肠炎恶化,但IEC来源的TNF并不起重要作用。
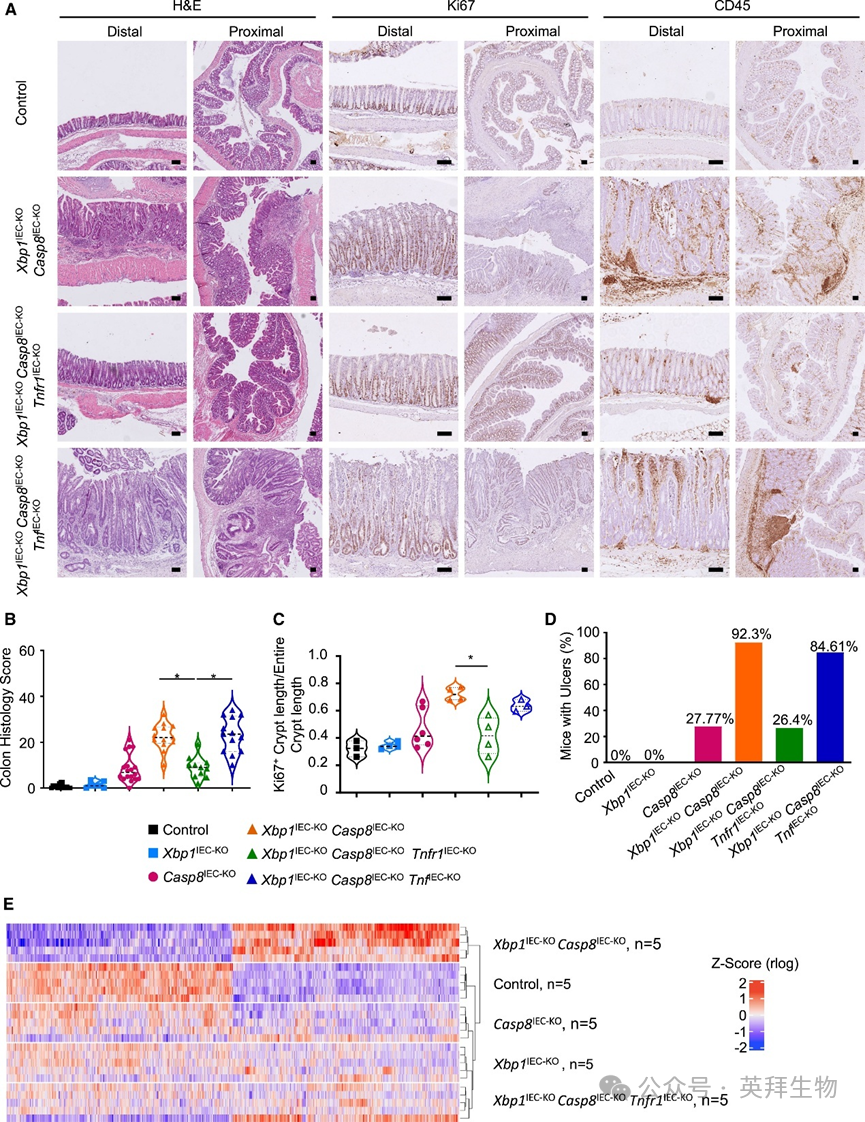
图3 IECs中TNFR1信号加重Xbp1IEC-KOCasp8IEC-KO小鼠的结肠炎严重程度
5.IEC特异性XBP1缺失损害结肠黏液层形成
RNA-seq数据分析显示,Xbp1IEC-KO小鼠结肠中仅有的显著减少的转录本是Muc2(图4A)。RT-qPCR分析、免疫染色及特异性抗体免疫印迹均证实,与对照组相比,Xbp1IEC-KO小鼠结肠中MUC2 mRNA和蛋白表达降低(图4B)。Xbp1IEC-KO小鼠结肠杯状细胞数量未减少,但细胞体积较小且MUC2表达降低。MUC2是肠道中最丰富的凝胶形成型黏蛋白,也是黏液层的主要组成部分。阿尔新蓝- PAS(AB-PAS)染色显示,与同窝对照组相比,Xbp1IEC-KO小鼠结肠黏蛋白水平降低。RNA-seq数据分析表明,其他黏液相关基因在Xbp1IEC-KO小鼠结肠中未见显著改变。MUC2是结肠上皮表面不可穿透黏液层的关键组分,能阻止管腔细菌与肠上皮细胞接触。我们推测IEC特异性XBP1缺失导致的Muc2表达减少可能损害黏液层形成,使得更多细菌接触上皮,从而加剧Xbp1IEC-KOCasp8IEC-KO和Xbp1IEC-KOFaddIEC-KO小鼠的结肠炎发展。为研究MUC2产生减少是否会破坏Xbp1IEC-KO小鼠保护性黏液层的形成,我们检测了用卡诺固定液(可保留黏液结构)固定的含粪便物质远端结肠组织切片。AB-PAS染色及MUC2免疫染色显示对照组小鼠结肠存在完整厚黏液层(图4C)。与之形成鲜明对比的是,Xbp1IEC-KO小鼠结肠黏液层厚度显著减少,部分区域完全缺失黏液层(图4C)。为量化比较黏液层厚度,我们测量了黏液总面积并将其与各小鼠结肠上皮周缘长度标准化。分析表明IEC特异性XBP1缺失导致整体黏液层厚度显著减少(图4D)。为评估Xbp1IEC-KO小鼠结肠中细菌与上皮表面的分离是否受损,我们采用真细菌核糖体RNA特异性探针(EUB338)进行荧光原位杂交(FISH),随后利用荆豆凝集素I(UEA-I)对α-连接岩藻糖残基进行免疫荧光染色以显示黏液层。对照组小鼠中细菌与结肠上皮明显分离,而Xbp1IEC-KO小鼠结肠中细菌侵入受损黏液层并与肠上皮细胞表面接触(图4E)。这些结果表明,IEC特异性XBP1缺失导致MUC2表达减少并损害保护性黏液层形成,使管腔细菌得以接触结肠上皮。
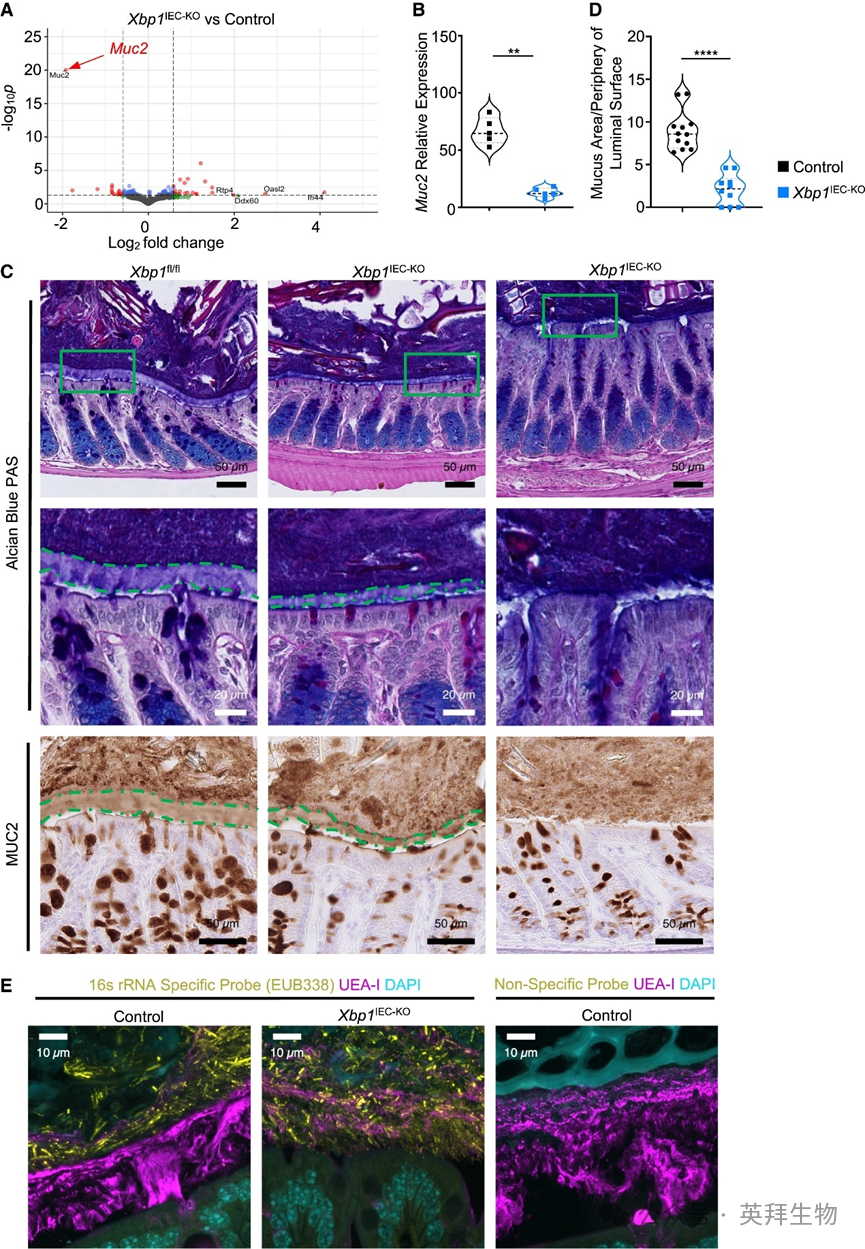
图4 IECs中XBP1表达调控结肠MUC2表达及黏液层形成
6.Xbp1IEC-KOCasp8IEC-KO小鼠出现细菌易位至结肠黏膜和全身性炎症
我们推测XBP1缺失引起的黏液层形成受损, combined with caspase-8缺失IECs坏死性凋亡所致上皮屏障破坏,可使细菌侵入黏膜和黏膜下层,导致Xbp1IEC-KOCasp8IEC-KO小鼠发生严重结肠炎。首先我们证实Xbp1IEC-KOCasp8IEC-KO小鼠也表现出MUC2表达降低和总黏蛋白水平减少(图5A)。与既往报道称IEC特异性caspase-8缺失降低结肠MUC2表达相反,我们的RNA-seq、RT-qPCR分析、MUC2免疫染色及AB-PAS染色均未发现Casp8IEC-KO小鼠结肠MUC2表达或黏液水平较对照组降低(图5A)。因此Xbp1IEC-KOCasp8IEC-KO小鼠结肠黏蛋白水平降低源于XBP1而非caspase-8缺失。为评估细菌向上皮下层易位情况,我们采用FISH技术对Casp8IEC-KO和Xbp1IEC-KOCasp8IEC-KO小鼠瑞士卷结肠切片进行细菌16s rRNA染色。果然,在几乎所有Xbp1IEC-KOCasp8IEC-KO小鼠(11只中10只)的黏膜下层检测到细菌存在,而Casp8IEC-KO小鼠仅37.5%(24只中9只)出现此现象(图5B)。此外,Xbp1IEC-KOCasp8IEC-KO小鼠(而非Casp8IEC-KO小鼠)出现脾脏肿大和外周血粒细胞、单核细胞数量增加(图5C和5D),表明细菌向上皮下层易位引发了全身免疫激活。这些结果证明,IEC特异性XBP1与caspase-8联合缺失导致细菌从管腔易位至结肠上皮下层并引发全身性炎症。
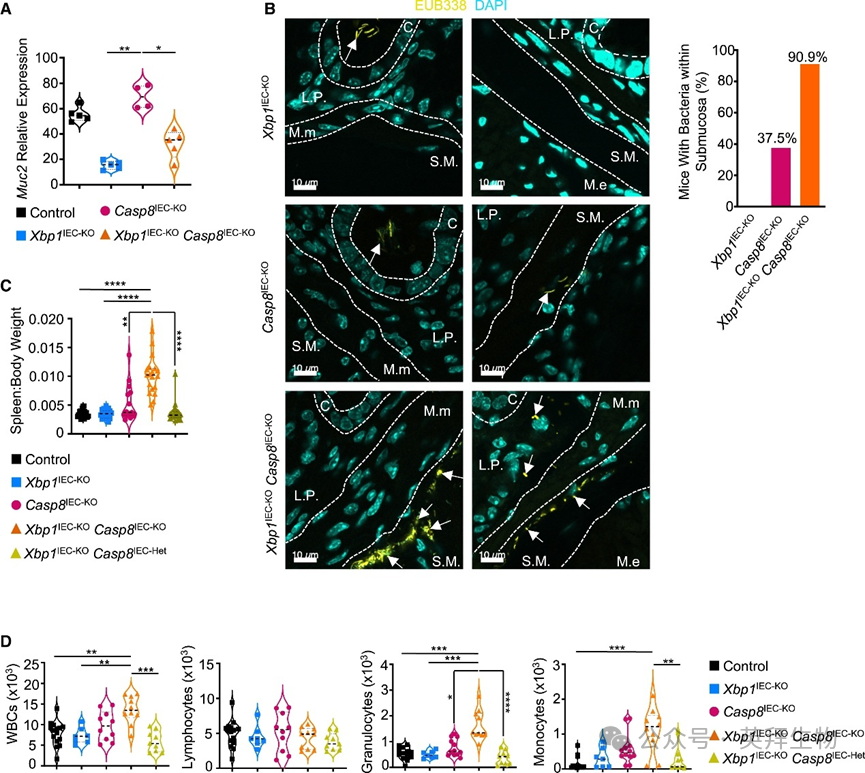
图5 上皮特异性XBP1抑制细菌易位并阻止Casp8IEC-KO小鼠全身性炎症
7.抑制坏死性凋亡可阻止Xbp1IEC-KOCasp8IEC-KO小鼠的细菌易位和全身性炎症
MUC2仅由结肠上皮杯状细胞产生和分泌。我们探究XBP1缺失是否可能诱导杯状细胞死亡,从而间接导致MUC2表达减少和黏液层受损。为此我们通过CC3免疫染色和TUNEL染色评估Xbp1IEC-KO与对照组小鼠结肠细胞死亡情况。未检测到Xbp1IEC-KO小鼠结肠死亡细胞数量增加,不支持XBP1缺失导致杯状细胞死亡的假设。我们随后检验联合抑制caspase-8诱导的凋亡和RIPK3-MLKL介导的坏死性凋亡能否挽救IEC特异性XBP1缺失导致的MUC2表达受损和黏液层形成障碍。我们对Xbp1IEC-KOCasp8IEC-KORipk3−/−和Xbp1IEC-KOCasp8IEC-KOMlkl−/−小鼠结肠切片进行MUC2免疫染色与AB-PAS染色,并对IECs中MUC2表达进行免疫印迹分析。这些实验表明联合抑制凋亡和坏死性凋亡未能挽救Xbp1IEC-KO小鼠的MUC2表达减少。此外,结肠横截面黏液层可视化与量化显示,Xbp1IEC-KOCasp8IEC-KORipk3−/−和Xbp1IEC-KOCasp8IEC-KOMlkl−/−小鼠的黏液层形成缺陷与Xbp1IEC-KO小鼠相似(图6A和6B)。这些结果表明Xbp1IEC-KO小鼠黏蛋白水平降低和黏液层形成受损并非由杯状细胞坏死性凋亡和/或凋亡增加引起。
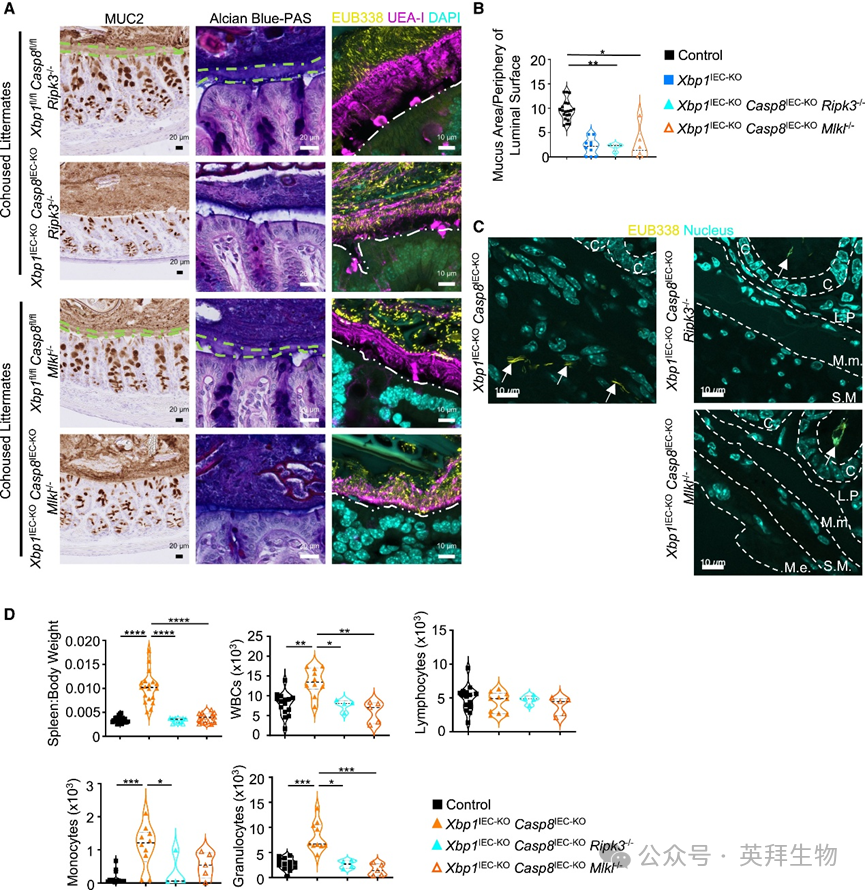
图6 上皮特异性XBP1以不依赖内在凋亡和坏死性凋亡的方式调控Muc2表达
我们随后评估抑制坏死性凋亡能否阻止细菌侵入Xbp1IEC-KOCasp8IEC-KORipk3−/−和Xbp1IEC-KOCasp8IEC-KOMlkl−/−小鼠黏膜和黏膜下层。与MUC2表达减少一致,我们发现细菌可侵入受损黏液层并抵达Xbp1IEC-KOCasp8IEC-KORipk3−/−和Xbp1IEC-KOCasp8IEC-KOMlkl−/−小鼠结肠上皮表面(图6A)。但结肠瑞士卷切片16s rRNA FISH染色显示,细菌未浸润Xbp1IEC-KOCasp8IEC-KORipk3−/−和Xbp1IEC-KOCasp8IEC-KOMlkl−/−小鼠结肠黏膜下层(图6C)。与无细菌浸润一致,Xbp1IEC-KOCasp8IEC-KORipk3−/−和Xbp1IEC-KOCasp8IEC-KOMlkl−/−小鼠未出现全身性炎症迹象,表现为脾脏大小正常且外周血粒细胞、单核细胞数量与同窝对照组相当(图6D)。这些结果表明,抑制坏死性凋亡虽不能挽救黏液层缺陷,但通过修复上皮屏障阻止了细菌向上皮下层浸润。
8.XBP1缺失以杯状细胞内在方式减少Muc2表达
肠道微生物群的存在与组成调节结肠MUC2产生及黏液层通透性和厚度。因此我们探究XBP1缺失是以细胞内在方式还是通过影响微生物群等其他因素间接导致Muc2表达减少。为解答此问题,我们培养对照组和Xbp1IEC-KO小鼠结肠类器官,并通过γ-分泌酶抑制剂DAPT抑制Notch信号诱导其分化为杯状细胞(图7A)。RT-qPCR分析显示DAPT处理强烈诱导对照组结肠类器官Muc2表达,而Xbp1IEC-KO小鼠类器官中Muc2表达显著降低(图7B)。XBP1缺失不影响杯状细胞分化,表现为对照组与XBP1缺失类器官中分泌细胞命运决定关键转录因子(包括Atoh1(Math1)、Spdef和Klf4)表达水平相当(图7B)。这些发现与Xbp1IEC-KO小鼠结肠杯状细胞数量未改变一致,且RNA-seq分析显示Xbp1IEC-KO小鼠结肠杯状细胞分化基因未减少(图4A)。因此XBP1缺失以细胞内在方式抑制杯状细胞MUC2表达。
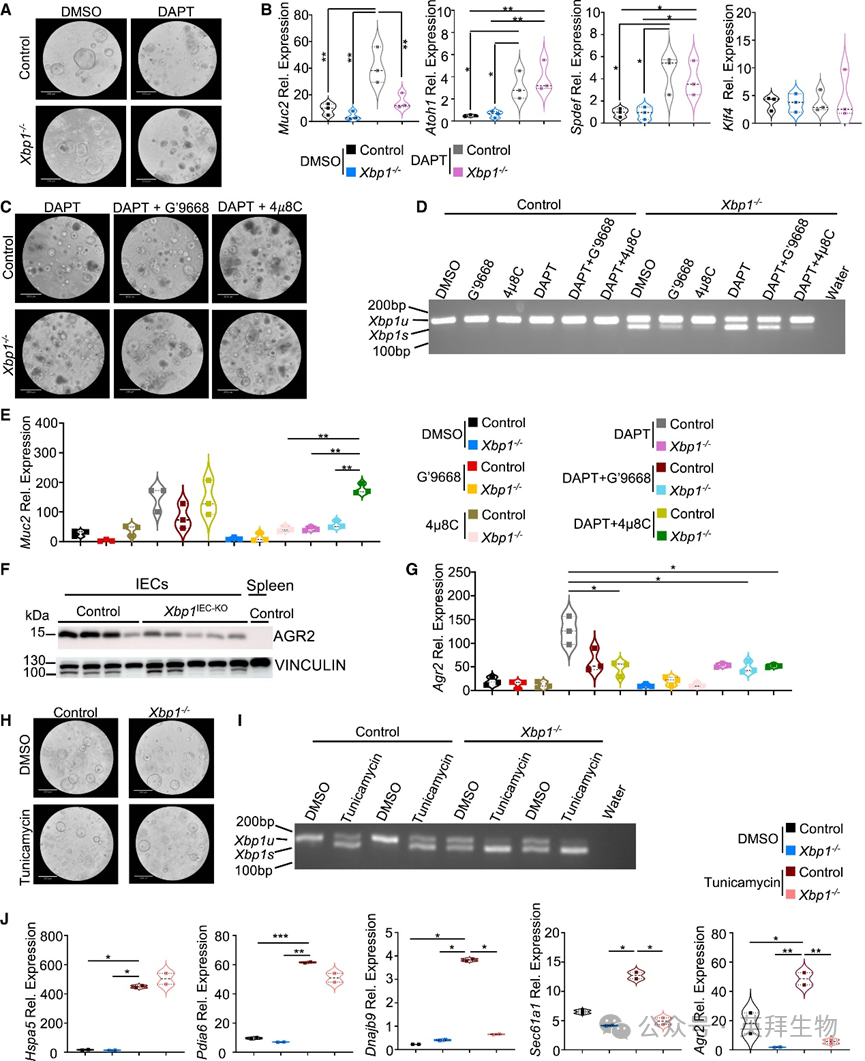
图7 XBP1以杯状细胞内在方式调控Muc2表达
IEC特异性XBP1缺失导致需肌醇酶1α(IRE1α)激活,表现为与对照组相比Xbp1IEC-KO小鼠结肠IECs中IRE1α磷酸化增加,这与既往报道一致。IRE1α自磷酸化诱导其核糖核酸酶结构域介导的RNase活性,从而介导未剪接Xbp1(Xbp1u)的非经典剪接,产生剪接型Xbp1(Xbp1s)。除Xbp1u的非经典剪接外,IRE1α还能通过其核酸内切酶结构域降解特定mRNA池,该过程称为 regulated IRE1-dependent decay(RIDD)。因此我们推测Muc2可能是XBP1缺失杯状细胞中活性IRE1α通过RIDD降解的靶标。为验证该假设,我们用IRE1α高特异性激酶抑制剂G′9668处理对照组或Xbp1IEC-KO小鼠类器官(图7C)。Cre介导的Xbp1基因loxP侧翼外显子2切除导致移码突变并引入提前终止密码子,使XBP1S蛋白表达缺失。由于外显子2缺失后IRE1介导的剪接位点保留且XBP1S缺失诱导IRE1激活,我们可通过Xbp1剪接实验评估对照组或Xbp1IEC-KO小鼠类器官中IRE1活性(图7D)。RT-PCR检测Xbp1u和Xbp1s显示XBP1缺失类器官中Xbp1剪接增加,而IRE1激酶抑制剂G′9668可抑制此现象(图7D)。但RT-qPCR分析表明缺失XBP1的杯状细胞分化类器官中Muc2 mRNA表达在G′9668处理后未增加(图7E)。除IRE1α外,黏膜上皮细胞表达IRE1旁系同源蛋白IRE1β(基因名ERN2)。IRE1β在杯状细胞中高度富集,对维持黏膜稳态起重要作用。与IRE1α相似,IRE1β含核酸内切酶结构域,可执行Xbp1u的非经典剪接。有观点认为IRE1β的核酸内切酶活性可降解Muc2 mRNA并参与肠道黏蛋白稳态。在Xbp1剪接实验中,我们检测到IRE1α激酶抑制剂G′9668存在时仍有残留Xbp1s条带(图7D)。因此我们探究XBP1缺失的杯状细胞中IRE1β是否可能被激活并降解Muc2 mRNA。为此我们用IRE1核酸内切酶抑制剂4μ8C(可抑制IRE1α和IRE1β的核酸内切酶活性)处理杯状细胞分化的对照组或Xbp1−/−类器官。确实,4μ8C处理抑制了IRE1激活(通过XBP1缺失类器官中Xbp1剪接检测证实)(图7D)。此外,4μ8C处理使Xbp1−/−类器官Muc2 mRNA表达恢复至对照组水平(图7E)。IRE1抑制不改变DAPT诱导的结肠类器官杯状细胞分化。这些结果支持Muc2在缺失XBP1的杯状细胞中被IRE1β降解。
前梯度蛋白同源物2(AGR2)属于蛋白质二硫键异构酶(PDI)家族,与炎症性肠病(IBD)存在关联。Agr2−/−小鼠表现为上皮内质网应激、随年龄进展的直肠脱垂,以及右旋葡聚糖硫酸钠(DSS)诱导的急性重症结肠炎易感性。AGR2在杯状细胞中高度富集表达,其缺失会导致杯状细胞MUC2产生减少和结肠黏液层形成受损,表明AGR2在维持肠道上皮内质网稳态和MUC2生物合成中起关键作用。此外,研究提出XBP1可调控肺上皮细胞中Agr2表达。AGR2与IRE1β相互作用,阻止其激活以及IRE1β介导的Xbp1u剪接。因此我们推测XBP1缺陷的杯状细胞可能表现出AGR2表达受损,导致IRE1β激活进而引起Muc2 mRNA降解。确实,免疫印迹分析显示与对照组相比,Xbp1IEC-KO小鼠IECs中AGR2表达减少(图7F)。与此一致,缺失XBP1的杯状细胞分化类器官中Agr2 mRNA表达较对照组降低(图7G)。虽然G′9668和4μ8C处理未改变Xbp1−/−类器官中Agr2表达,但抑制IRE1α激酶活性或IRE1α/IRE1β核酸内切酶活性会降低对照组类器官的Agr2表达(图7G),表明剪接型XBP1可诱导Agr2表达。与各自对照组相比,XBP1缺陷的IECs或Xbp1−/−类器官中IRE1β表达未受影响。为研究XBP1是否调控AGR2表达,我们采用衣霉素孵育类器官化学诱导内质网应激(图7H)。Xbp1u剪接增加以及Hspa5(免疫球蛋白结合蛋白[BiP])mRNA表达升高证实衣霉素处理成功诱导类器官内质网应激(图7I和7J)。此外,RT-qPCR分析显示对照组和XBP1缺陷类器官中Pdia6(激活转录因子6[ATF6]介导的UPR靶基因)被强烈诱导(图7J)。但缺失XBP1的类器官未能上调两个已知XBP1靶基因Dnajb9和Sec61a1的表达(图7J)。而且与对照组类器官相反,衣霉素处理未能增加Xbp1−/−类器官中Agr2表达(图7J)。这些结果表明XBP1通过诱导AGR2表达阻止IRE1β激活并抑制IRE1β介导的Muc2 mRNA降解。
结论
我们的研究揭示了IECs中内质网应激与坏死性凋亡调控之间存在重要交互作用,共同维持健康的免疫稳态并预防肠道炎症。这些发现暗示不同通路间的功能冗余可能是大多数IBD易感基因外显率低的基础,并表明可能需同时影响肠道屏障不同组分的联合缺陷才能启动肠道炎症并使其慢性化。
参考文献:
Kaya GG, Schwarzer R, Dannappel M, Vlantis K, Göbel U, Kondylis V, Nedospasov SA, Pasparakis M. Unfolded protein response transcription factor XBP1 suppresses necroptosis-induced colitis by reinforcing the mucus barrier. Immunity. 2025 Aug 22:S1074-7613(25)00332-2. doi: 10.1016/j.immuni.2025.07.023. Epub ahead of print. PMID: 40865520.