






精氨酸酶1会促使线粒体嵴发生重塑,并引发缺血/缺氧所致血管功能障碍中的程序性细胞死亡
缺血/缺氧损伤显著损害血管功能,对患者预后产生不利影响。线粒体结构与功能的变化与缺血/缺氧诱导的血管功能障碍密切相关。该过程的机制仍不清楚。使用大鼠缺血模型和缺氧血管平滑肌细胞(VSMCs),结合透射电子显微镜、超分辨率显微镜和代谢分析来分析线粒体嵴的结构与功能变化。多组学方法揭示缺血VSMCs中精氨酸酶1(Arg1)上调,通过体内外敲除模型证实Arg1缺失可保护线粒体嵴、线粒体与血管功能,并限制mtDNA释放。机制上,Arg1与Mic10相互作用导致线粒体嵴重塑,同时缺氧诱导的VDAC1乳酸化导致mPTP开放及VSMCs mtDNA释放。释放的mtDNA通过激活cGAS-STING通路引发VSMCs PANoptosis。ChIP-qPCR结果显示乳酸介导的Arg1上调由H3K18la升高所致。靶向VSMCs的纳米材料PLGA-PEI-siRNA@PM-α-SMA(NPsiArg1)显著改善血管功能障碍。本研究揭示缺血/缺氧后血管功能障碍的新机制:乳酸调控的Arg1表达在细胞核与线粒体之间形成损伤性正反馈环,导致线粒体嵴紊乱与mtDNA释放,最终引发VSMCs PANoptosis。靶向抑制VSMCs Arg1为缓解缺血/缺氧性血管损伤提供潜在治疗策略。该研究于2025年5月发表于《Signal Transduction and Targeted Therapy》,题目为“Arginase 1 drives mitochondrial cristae remodeling and PANoptosis in ischemia/hypoxia-induced vascular dysfunction”,影响因子52.7。
技术路线

研究思路
1. 线粒体嵴损伤与缺血/缺氧诱导的血管功能障碍相关
与假手术组相比,缺血大鼠离体肠系膜上动脉对 norepinephrine的收缩反应显著减弱(图1a, b)。10⁻³ mol/L去norepinephrine刺激导致假手术组SMA直径明显缩小,而缺血组各剂量norepinephrine均无效(图1c, d)。缺氧VSMCs收缩力显著低于正常组(图1e, f)。缺血组24小时生存率与生存时间显著下降(补充图S1d, e)。缺氧VSMCs线粒体嵴结构紊乱、空泡增多、嵴频率降低(图1g, h),Hessian-SIM超分辨率显微镜结果与透射电镜一致(图1i, j)。缺氧组氧耗率、ATP降低,ROS升高,线粒体膜电位下降(图1k, l;补充图S1f–i)。缺氧VSMCs mtDNA释放显著增加(图1m–p)。NOS抑制剂L-NAME未引起线粒体功能障碍与mtDNA释放(补充图S1j–o)。
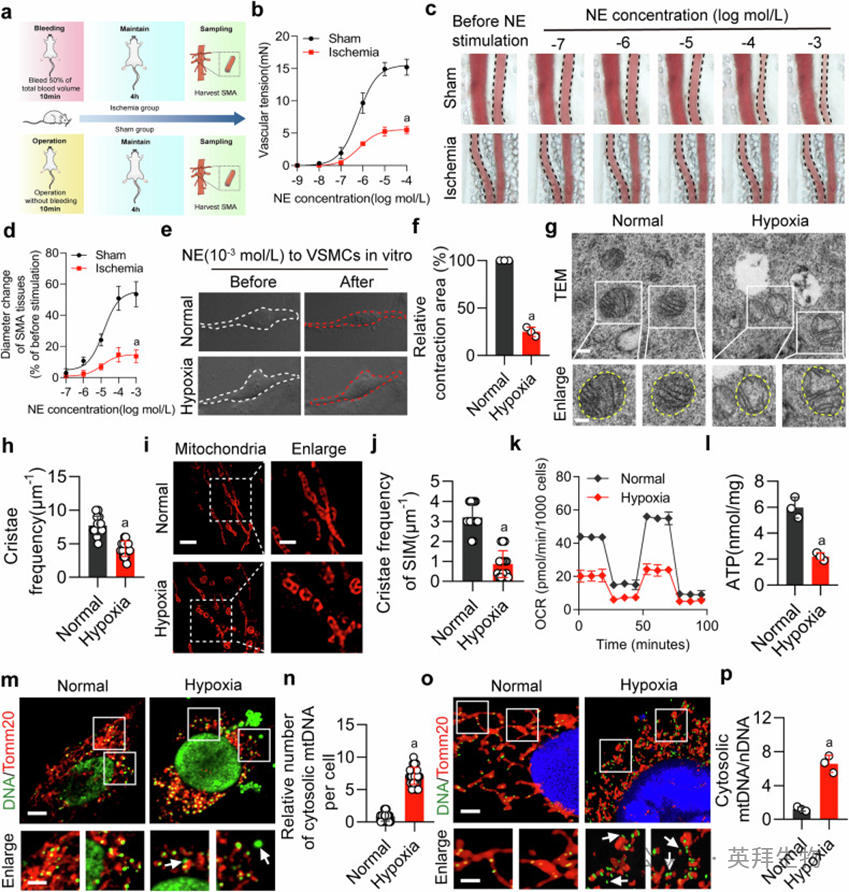
图1. 线粒体嵴损伤与缺血/缺氧诱导的血管功能障碍相关
2. 干预Arg1改善血管功能与线粒体嵴结构
GEO数据库失血性休克患者转录组(GSE64711)分析显示Arg1为休克组与对照组差异基因中在缺血小鼠VSMCs单细胞测序中上调的基因(图2a–c)。构建VSMCs条件性Arg1敲除小鼠与AAV介导Arg1敲低大鼠,证实缺血组Arg1表达升高,而敲除/敲低可恢复SMA收缩反应、增加肠系膜血流并提高生存率(图2f–i;补充图S2c–g)。体外实验显示缺氧VSMCs Arg1表达上调(图2j),siRNA干扰Arg1后VSMCs收缩力恢复(图2l, m)。Arg1敲低改善缺氧VSMCs线粒体嵴结构,增加嵴频率(图2n–q),抑制mtDNA释放,提升OCR、ATP及膜电位(图2r–v;补充图S3c–g)。Arg1酶活性非其调控作用关键(补充图S4c–f)。
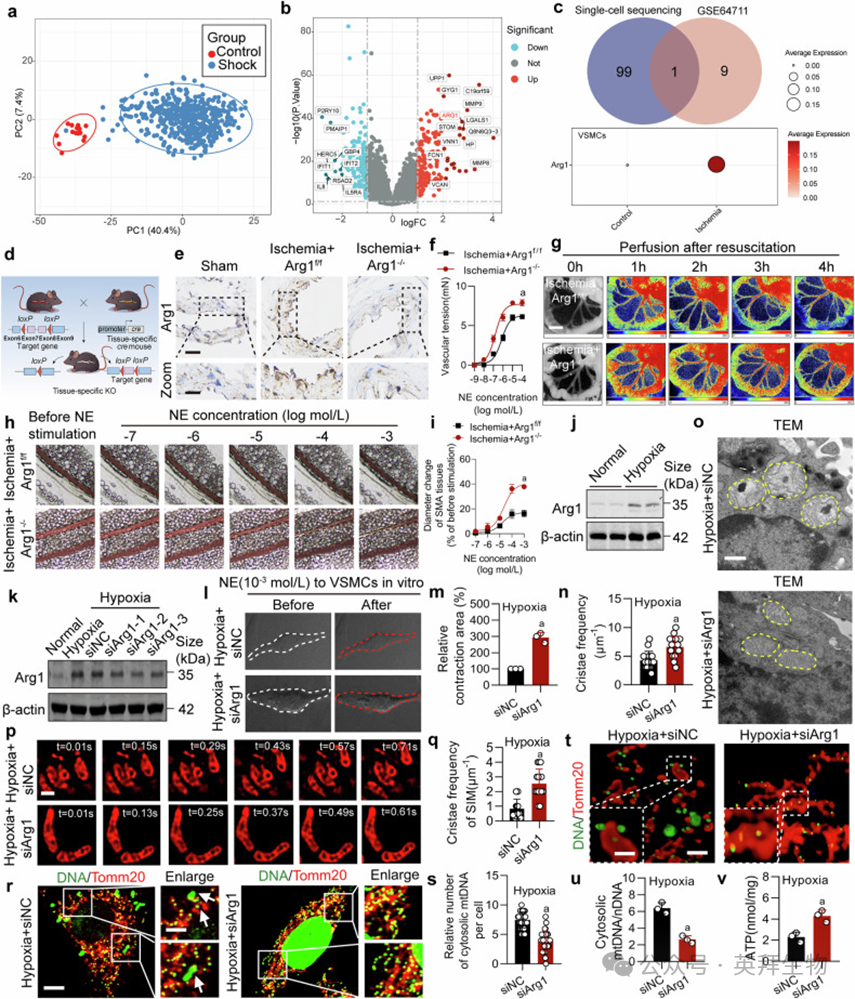
图2. 干预Arg1改善血管功能与线粒体嵴结构
3. Arg1与Mic10相互作用导致Mic10构象改变
共免疫沉淀-质谱鉴定292个Arg1互作蛋白,其中Mic10(MICOS复合物关键亚基)与Arg1结合(图3a–c)。分子对接显示Arg1 Glu42与Mic10结合最强(图3d)。缺氧增强Arg1-Mic10结合,Arg1干扰削弱其互作(图3e–g)。缺氧增加线粒体Arg1水平,siArg1降低其水平(图3h–k)。分子动力学模拟表明Arg1结合破坏Mic10寡聚化,增加Mic10体积与构象不稳定性(图3l–o;补充图S5)。
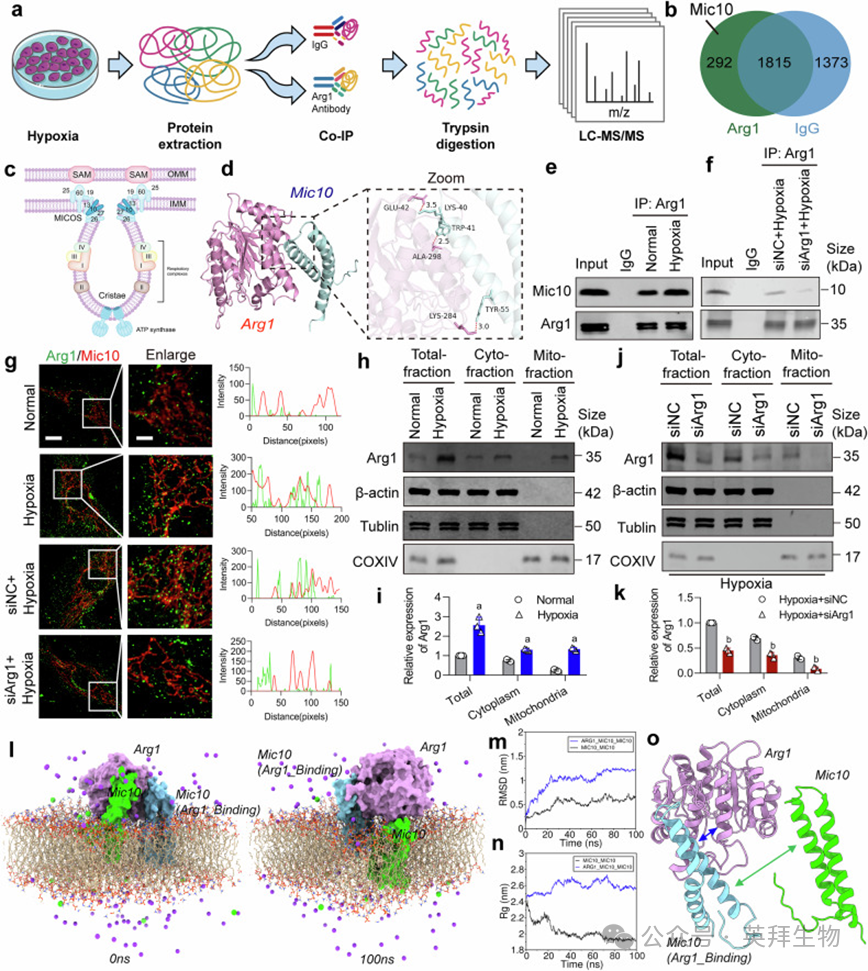
图3. Arg1与Mic10相互作用导致Mic10构象改变
4. Arg1 E42A突变逆转线粒体嵴紊乱
Arg1 E42A突变显著削弱Arg1与Mic10结合(图4a–c)。透射电镜与超分辨率显微镜显示E42A突变改善缺氧VSMCs线粒体嵴结构,减少空泡、增加嵴频率(图4d, e;补充图S6a, b)。E42A突变提升OCR、ATP,抑制ROS,降低mtDNA释放(图4f–o)。
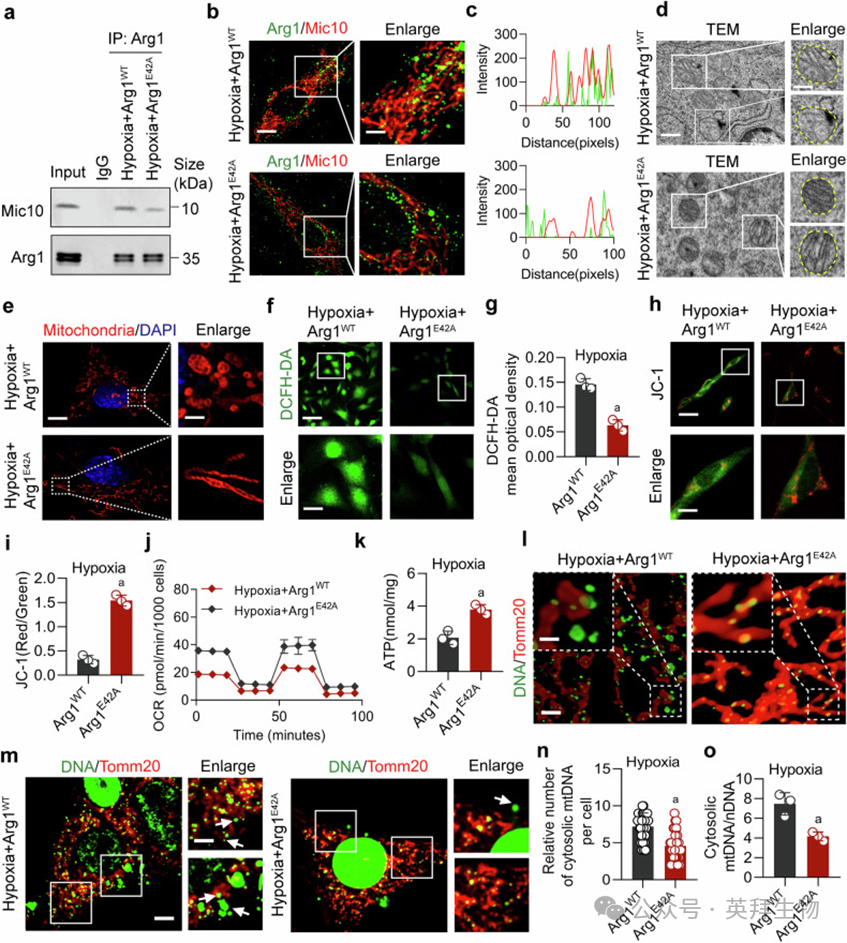
图4. Arg1 E42A突变逆转线粒体嵴紊乱
5. Arg1上调与VDAC1乳酸化共同介导mtDNA释放
缺氧导致mPTP开放,Arg1干扰或E42A突变抑制其开放(图5a, b)。缺氧诱导VDAC1 K224位点乳酸化(图5e–k),Arg1敲低通过改善线粒体功能抑制VDAC1乳酸化(图5h, l)。VDAC1 K224R突变抑制mtDNA释放(图5m)。过表达Arg1增加乳酸与mtDNA释放,K224R突变可逆转此效应(图5l, m)。
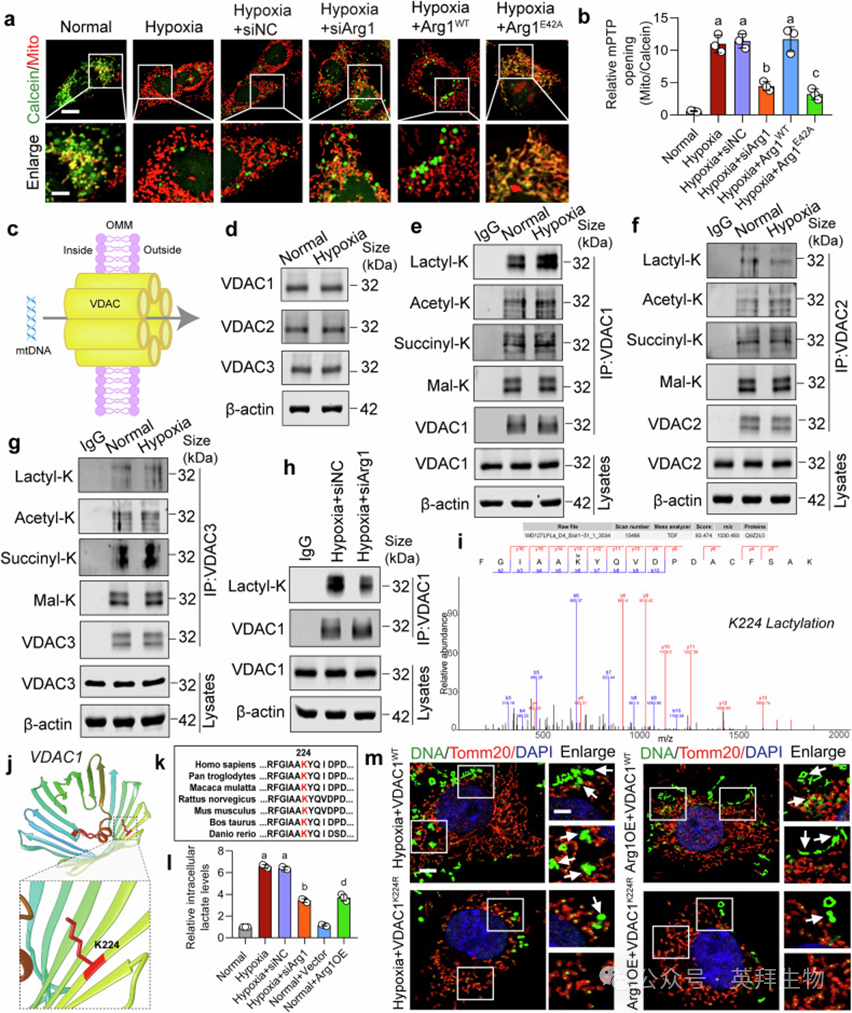
图5. Arg1上调与VDAC1乳酸化共同介导mtDNA释放
6. mtDNA释放经cGAS-STING通路触发VSMCs PANoptosis
缺氧VSMCs cGAS-STING通路激活,凋亡(Cleaved-Caspase-3、Bax)、焦亡(NLRP3、Caspase-1、GSDMD-N)、坏死性凋亡(p-RIPK3、p-MLKL)相关蛋白表达升高(图6a, b)。Arg1干扰、E42A突变或STING敲除抑制PANoptosis(图6c–h;补充图S8)。STING激动剂逆转Arg1敲除对缺血小鼠血管功能的保护作用(图6i–m)。
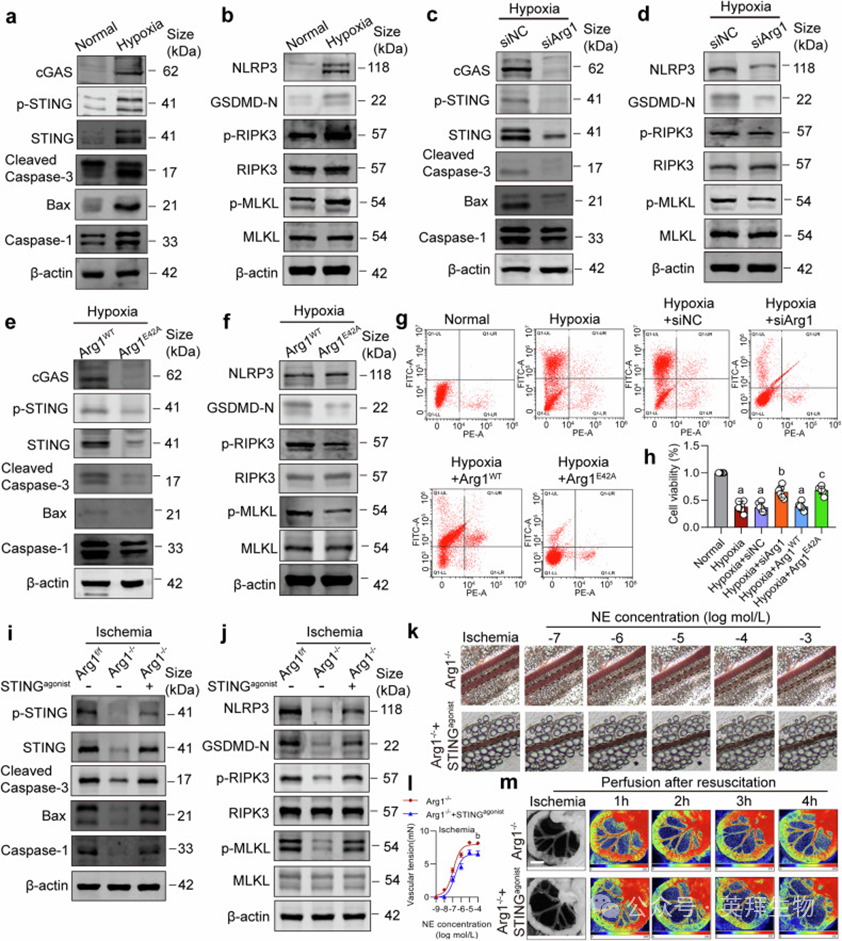
图6. mtDNA释放经cGAS-STING通路触发VSMCs PANoptosis
7. 乳酸通过H3K18la上调Arg1表达
缺氧VSMCs中乳酸水平与Arg1、H3K18la表达同步升高(图7a–c)。外源乳酸直接刺激VSMCs可剂量依赖地上调Arg1与H3K18la(图7d, e)。ChIP-qPCR显示H3K18la与Arg1启动子结合随缺氧时间延长而增强(图7f)。LDHA抑制剂Oxamate抑制乳酸生成后,H3K18la及Arg1表达下降(图7g–i),且H3K18la与Arg1启动子结合减少(图7j)。
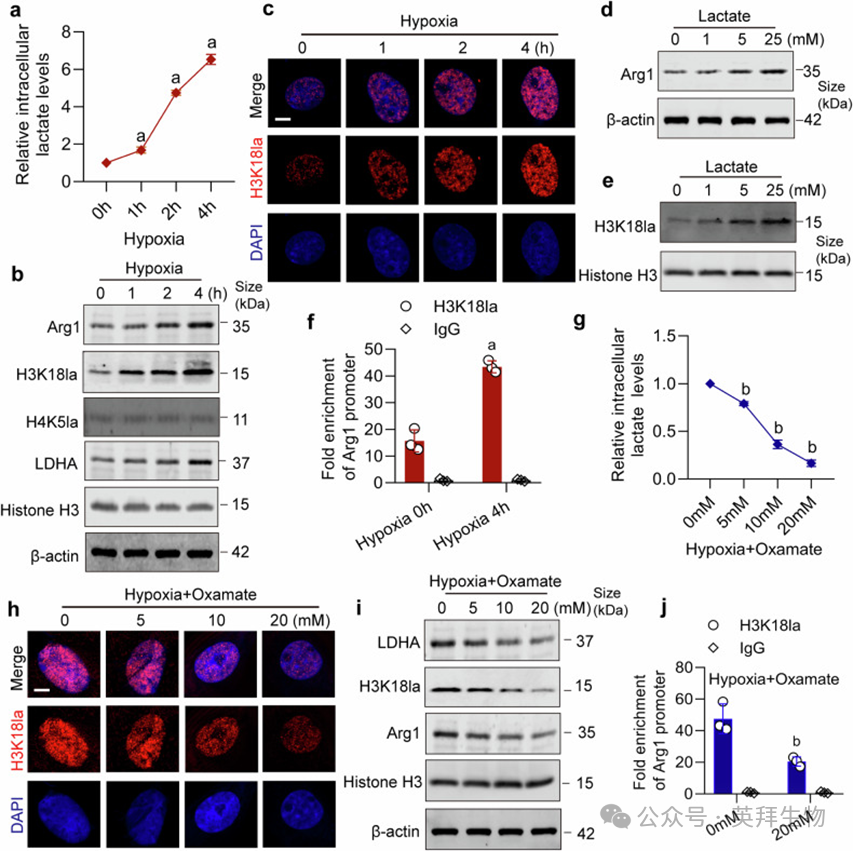
图7. 乳酸通过H3K18la上调Arg1表达7. 乳酸通过H3K18la上调Arg1表达
8. 构建靶向VSMCs的纳米颗粒NP-siArg1
以PLGA为核心,包覆血小板膜(PM)并偶联α-SMA抗体,制备PLGA-PEI-siRNA@PM-α-SMA(NP-siArg1)。粒径284.97 nm,Zeta电位9.98 mV(表1;图8c, d)。电镜显示PM成功包覆(图8e)。SDS-PAGE与WB验证膜蛋白与抗体存在(图8f, g)。体外释放:NP-siArg1 72 h累积释放95%,显著延缓siRNA突释(图8h)。细胞摄取实验证实NP-siArg1选择性进入VSMCs,缺氧条件下穿透VEC单层效率更高(图8i;补充图S11g–i)。
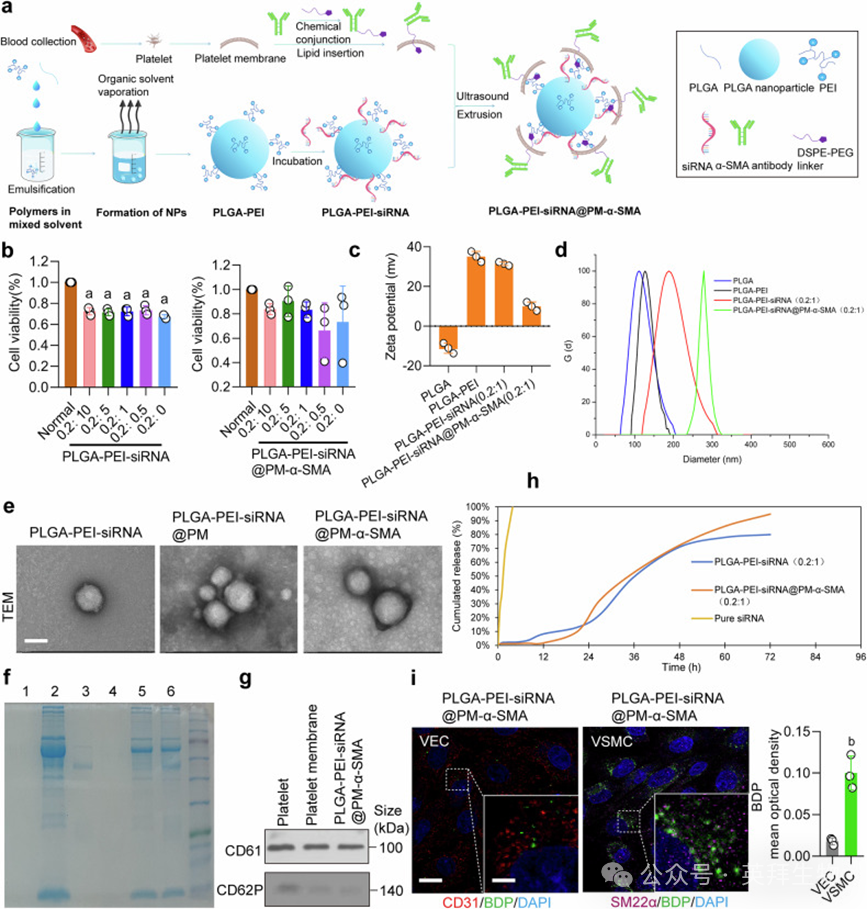
图8. 构建靶向VSMCs的纳米颗粒NP-siArg1
9. NP-siArg1治疗缺血/缺氧性血管功能障碍
NP-siArg1(3 mg/kg)较AAV-shArg1更显著下调缺血大鼠SMA中Arg1表达(图9a, b)。血管功能检测显示NP-siArg1恢复SMA对NE的收缩反应(图9c–e),提高生存率(补充图S12b, c)。缺氧VSMCs中,NP-siArg1较游离siArg1更明显增加线粒体嵴频率(图9f, g),降低ROS,提升ATP、OCR及膜电位(图9h–k),抑制mPTP开放及mtDNA释放(图9l–o),并减少PANoptosis相关蛋白表达(图9p)。
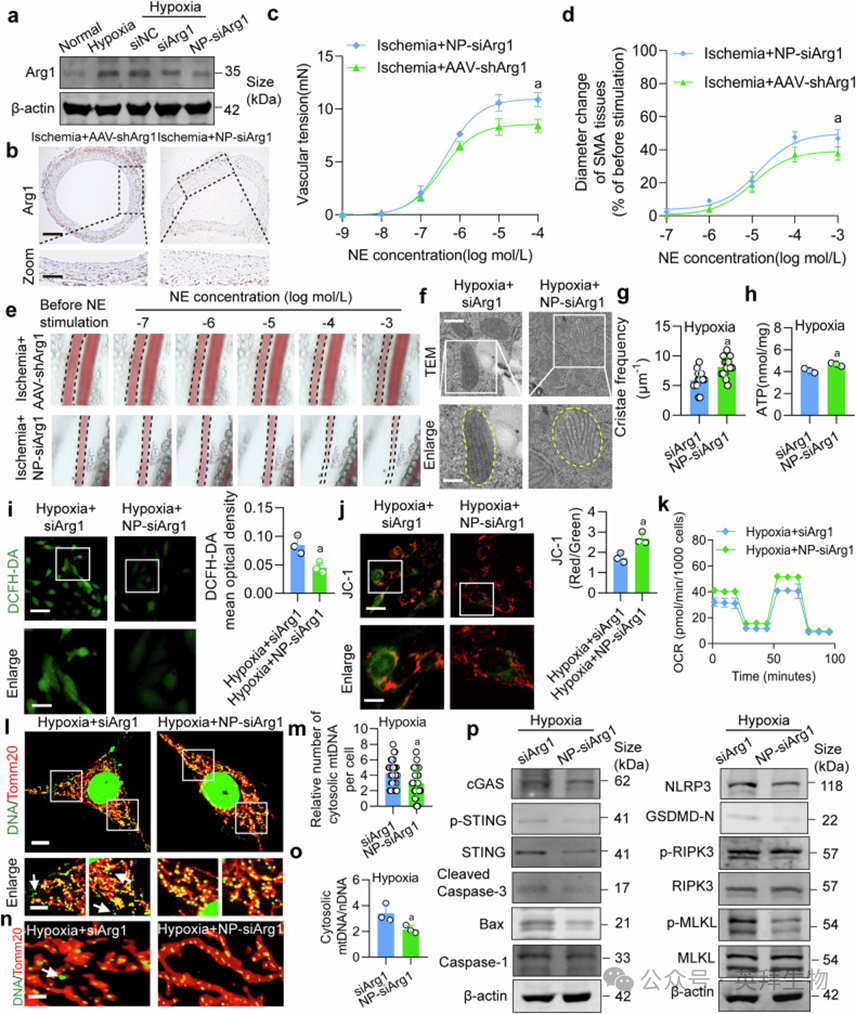
图9. NP-siArg1治疗缺血/缺氧性血管功能障碍
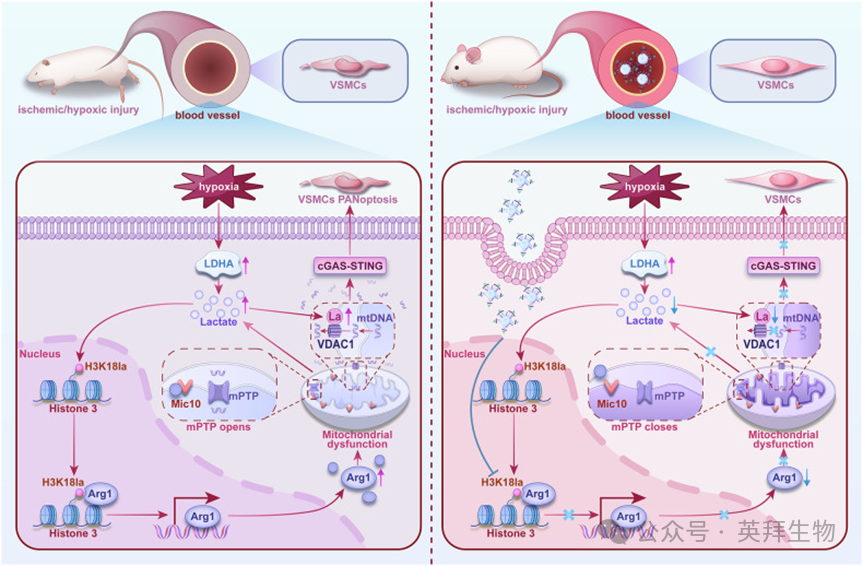
图10 图形摘要。
缺血/缺氧 → VSMCs乳酸积累 → 组蛋白H3K18乳酸化(H3K18la)↑ → 结合Arg1启动子 →Arg1表达上调。
Arg1进入线粒体,进一步加重线粒体功能障碍 → 乳酸产生↑(形成核-线粒体损伤循环)。
线粒体内膜:Arg1与Mic10蛋白结合 → 抑制Mic10寡聚化 → 线粒体嵴结构紊乱 → 线粒体膜通透性转换孔(mPTP)开放。
线粒体外膜:乳酸诱导VDAC1第224位赖氨酸乳酸化(VDAC1-K224la)→ 促进mtDNA外泄。
释放的mtDNA结合cGAS → 激活STING-TBK1通路 → 触发PANoptosis → 血管收缩功能丧失
纳米颗粒NP-siArg1:沉默VSMCs中Arg1表达 → 阻断线粒体嵴重构 → 改善血管功能。
参考文献
1. She H, Zheng J, Zhao G, Du Y, Tan L, Chen ZS, Wu Y, Li Y, Liu Y, Sun Y, Hu Y, Zuo D, Mao Q, Liu L, Li T. Arginase 1 drives mitochondrial cristae remodeling and PANoptosis in ischemia/hypoxia-induced vascular dysfunction. Signal Transduct Target Ther. 2025 May 28;10(1):167.