






WSTF核自噬调节慢性炎症而非急性炎症
急性炎症是作者身体用来对抗感染的一种基本反应。然而,在没有感染的情况下,慢性炎症可能在关节炎、癌症、自身免疫性疾病、代谢功能障碍相关脂肪性肝炎(MASH)以及大多数与衰老相关的病理等慢性疾病的发病和进展中起关键作用。区分慢性炎症与急性炎症的潜在机制尚不清楚,这给针对这些重大疾病的靶向疗法的开发带来了挑战。在此,作者发现了一种将这两种反应区分开来的机制:在慢性炎症而非急性炎症期间,染色质重塑受到核自噬的影响,其中ISWI染色质重塑复合体的WSTF蛋白与核内的ATG8自噬蛋白家族相互作用。这种相互作用导致WSTF从核内输出,随后在细胞质中被自噬体和溶酶体降解。WSTF的缺失会导致炎症基因上的染色质开放,从而放大炎症反应。能够阻断WSTF-ATG8相互作用的细胞穿透肽不会影响急性炎症,但能在衰老以及小鼠模型和患者样本的MASH和骨关节炎中抑制慢性炎症。在不削弱急性炎症的情况下特异性靶向慢性炎症的能力,为治疗常见的慢性炎症性疾病提供了一种方法。总之,作者的研究确定了WSTF核自噬是慢性炎症反应中的一个关键调节机制,通过稳定WSTF蛋白水平或阻断其与ATG8的相互作用,可特异性地抑制慢性炎症,为慢性炎症性疾病的治疗提供了新的策略和靶点。该研究于2025年7月发表在《Nature》,IF 48.5分。
技术路线:
主要研究结果:
1.筛选核自噬底物
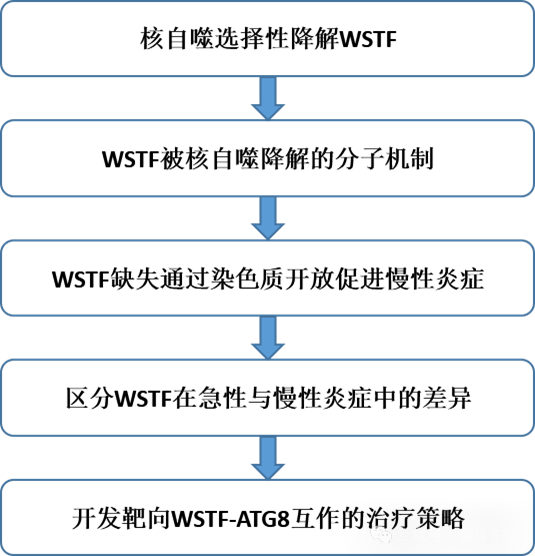
自噬蛋白的核内结合伴侣仍是待深入研究的领域。作者首先在人源二倍体成纤维细胞IMR90中稳定表达6种ATG8亚型(LC3A、LC3B、LC3C、GABARAP、GABARAPL1和GABARAPL2)(图1a)。随后采用能有效溶解染色质蛋白的核分离方案,进行免疫共沉淀(co-IP)联合串联质谱标签(TMT)质谱分析(图1a)。与已发表的聚焦细胞质互作的自噬互作网络相比,作者鉴定的大多数核内ATG8结合伴侣均为首次报道。这种差异可能源于细胞裂解方案的不同:既往互作组学研究多采用NP-40或Triton X-100裂解细胞,随后从上清液中进行co-IP,从而丢弃了沉淀中的染色质组分。
对核内ATG8结合伴侣进行基因本体(GO)分析显示,其显著富集于翻译、RNA加工,尤其是染色质重塑相关蛋白(图1b)。在GFP-LC3B转基因小鼠中也观察到类似模式:通过比较脑组织胞质与核组分中GFP的co-IP结果,发现染色质重塑蛋白与人和小鼠ATG8的结合(图1c,d)。染色质重塑蛋白通过调控核小体定位和染色质结构影响基因表达与沉默。
为深入筛选核自噬底物,作者研究了细胞衰老——一种细胞周期停滞状态,其中核自噬已在哺乳动物系统中被发现。衰老细胞在慢性疾病和老化组织中累积,并分泌多种促炎细胞因子、趋化因子、蛋白酶和生长因子,统称为衰老相关分泌表型(SASP)。由癌基因激活或亚致死剂量DNA损伤(图1e,f)诱导的衰老均显示染色质重塑蛋白的丢失(图1f)。通过交叉比对上述筛选中的染色质重塑蛋白,作者鉴定出WSTF为共同靶点(图1g)。WSTF与ATG8结合,并在衰老细胞中以ATG7依赖的方式下调;RNA测序(RNA-seq)数据显示其mRNA水平在衰老中保持不变。
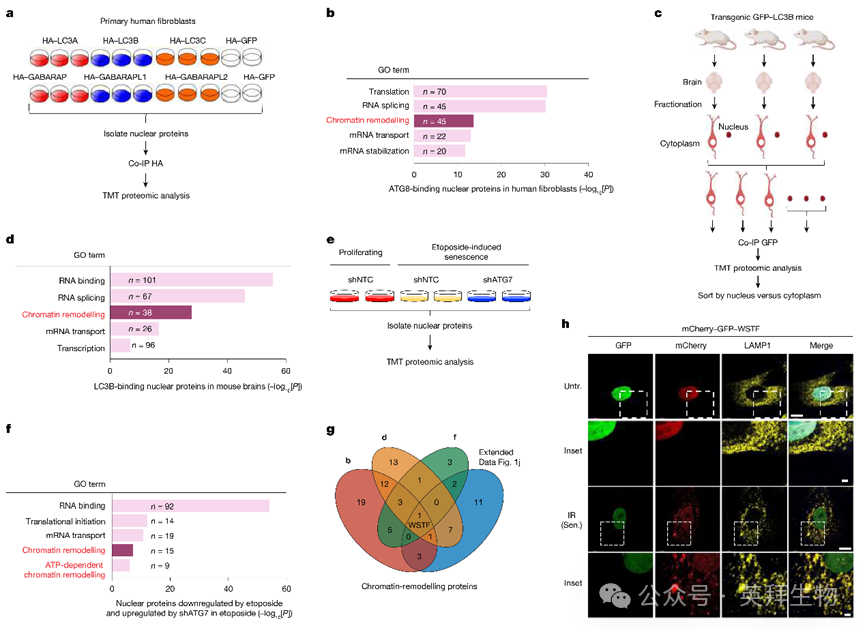
图1:核自噬以染色质重塑蛋白WSTF为靶点
2.核自噬降解WSTF
WSTF(又称BAZ1B;下文统称WSTF)是ISWI染色质重塑复合体的亚基,通过形成有序的核小体阵列将染色质从开放状态转变为闭合状态,从而抑制基因表达。WSTF与ISWI复合体的ATP酶SNF2H(又称SMARCA5)结合,后者沿DNA滑动核小体。SNF2H的染色质定位与催化活性受其互作伴侣(包括WSTF)调控。
多种细胞类型中,由不同方式诱导的衰老均导致WSTF蛋白减少(,而在静止或饥饿状态下其水平不变。SNF2H在衰老中基本不变。尽管WSTF蛋白水平在衰老中持续降低,其mRNA水平无变化。自噬基因(如ATG7、ATG13或RB1CC1/FIP200)失活或溶酶体抑制剂bafilomycin A1处理可抑制衰老中WSTF蛋白的丢失,而蛋白酶体抑制剂无此作用。此外,衰老过程中WSTF从核转移至胞质,并与溶酶体标志物LAMP1共定位,这一现象在内源蛋白和mCherry-GFP-WSTF报告系统中均得到验证(图1h),红色信号无绿色信号表明融合蛋白处于酸性环境。相反,SNF2H在衰老中仍滞留于核内。
3. WSTF自噬降解的分子机制
作者进一步发现GABARAP是直接结合WSTF(而非SNF2H)的主要ATG8亚型。双分子荧光互补实验显示,基础状态下GABARAP-WSTF互作发生于核内,而衰老中胞质内检测到互作,并与自噬体和溶酶体共定位。GABARAP的Phe77残基和WSTF的474-483区域(尤其是Leu476)对互作至关重要。无法结合GABARAP的WSTF突变体在衰老中自噬降解受损。这些结果共同证实WSTF是核自噬的底物。
4. ATM促进WSTF降解
为探究衰老中WSTF选择性降解的机制,作者关注GABARAP-WSTF互作,其以磷酸化依赖的方式在衰老中显著增强。既往研究表明ATM激酶驱动SASP25,其在衰老中被激活,但在饥饿或静止中不激活26。ATM抑制剂KU-55933(ATMi)处理不仅减少GABARAP-WSTF结合,还抑制衰老细胞中WSTF的下调。ATMi同样抑制WSTF的核-胞质穿梭与降解。靶向ATM的短发夹RNA(shRNA)进一步证实其介导GABARAP-WSTF结合与WSTF下调的作用。
5. WSTF缺失促进SASP
为探究WSTF缺失的意义,作者过表达WSTF并检测衰老关键特征。WSTF过表达对衰老相关β-半乳糖苷酶(SA-β-gal)及衰老标志物lamin B1和p16INK4a(p16)影响甚微,但显著抑制SASP(通过免疫印迹、RT-qPCR和细胞因子芯片检测)。WSTF失活则增强SASP基因表达。RNA-seq进一步证实WSTF对SASP的调控作用(图2a)。此外,在已建立且丢失WSTF的衰老细胞中强制表达WSTF可抑制SASP。机制上,WSTF不影响p38MAPK、DNA损伤反应、mTOR、胞质染色质片段(CCF)、cGAS-STING通路、NF-κB核转位或衰老相关异染色质灶,提示其通过未知机制抑制SASP。
作者进一步分别敲除WSTF的功能域,包括具酪氨酸激酶活性的WAC结构域27、结合SNF2H(ISWI复合体ATP酶)的BAZ1/2结构域,以及结合组蛋白的PHD和溴结构域(BRO)。BAZ1/2结构域和PHD-BRO结构域对WSTF抑制SASP至关重要,而WAC结构域非必需。此外,SNF2H失活阻断WSTF抑制SASP的能力,提示SNF2H结合及染色质关联对WSTF抑制SASP至关重要。作者因此通过转座酶可及性染色质测序(ATAC-seq)检测WSTF是否调控SASP基因的染色质可及性。衰老诱导导致SASP基因染色质可及性显著增加,而WSTF强制表达抑制这种增加(代表性示例见图2b)。差异可及区域分析显示促炎基因显著富集。通过分析WSTF在衰老中下调基因关联的转录因子,作者发现NF-κB及其RELA(p65)亚基显著富集,后者是包括大多数SASP基因在内的多种促炎基因的关键转录因子。与此一致,p65直接与WSTF(而非SNF2H)结合。因此,WSTF与NF-κB结合并抑制其靶位点的染色质可及性,从而抑制促炎基因表达。这些结果不排除WSTF通过其他机制间接抑制促炎基因表达的可能性,作者将在讨论部分进一步探讨。
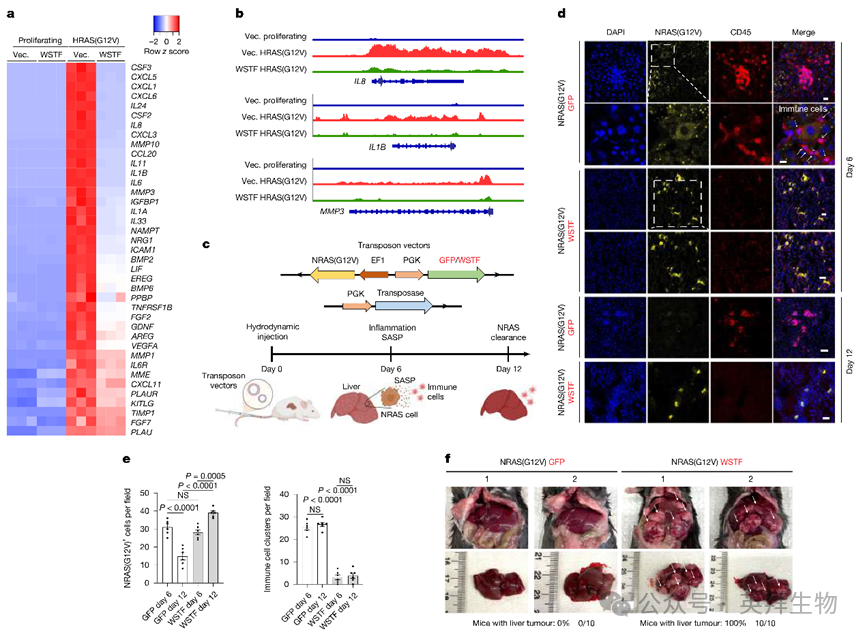
图2:WSTF的缺失会促进衰老和癌症的炎症
6. WSTF缺失在体内驱动炎症
接下来,作者利用小鼠模型研究WSTF与体内炎症的关系。通过水动力尾静脉注射在肝细胞中表达NRAS(G12V),诱导细胞衰老、SASP以及癌前细胞的免疫监视30(图2c)。NRAS(G12V)在肝细胞中的表达导致WSTF的丢失。因此,作者在同一载体中同时表达WSTF和NRAS(G12V)(图2c)。第6天时,NRAS(G12V)-GFP小鼠显示CD45阳性免疫细胞浸润NRAS(G12V)阳性肝细胞,而NRAS(G12V)-WSTF小鼠的免疫细胞浸润减少(图2d,e),伴随肝脏促炎基因和免疫细胞基因表达受损。第12天时,对照组中NRAS(G12V)阳性肝细胞减少,而WSTF组未减少(图2d,e)。NRAS(G12V)阳性肝细胞清除受损可导致表达NRAS(G12V)的肝肿瘤形成21,30。6个月后,对照组小鼠无一发生肝肿瘤(0/10),而WSTF组所有小鼠均出现严重的肝内肿瘤(10/10)(图2f)。综上,这些数据强烈表明WSTF的丢失是诱导小鼠对致癌RAS产生炎症和免疫监视的关键事件。
上述小鼠数据促使作者研究癌症,癌症常与慢性促炎基因表达相关。作者分析了癌症细胞系百科全书(CCLE)数据库的蛋白质组资源,该数据库包含300多种癌症细胞系的蛋白质组信息31。将WSTF表达最低和最高的50%细胞系分组,比较WSTFlow和WSTFhigh亚组中关键促炎基因的表达。结果显示,较高的WSTF蛋白表达与较高的SNF2H表达和较低的促炎基因表达相关。实验上,shRNA介导的WSTF失活增加了多种癌症细胞系中IL6和IL8的表达。综上,作者得出结论:WSTF的缺失在衰老和癌症中促进炎症。
7. WSTF在慢性与急性炎症中的作用
在揭示WSTF在衰老和癌症(均为慢性疾病)中的炎症作用后,作者进一步研究其在急性和慢性炎症中的行为。双链DNA(dsDNA;ISD)或双链RNA(poly(I:C))转染、IFNβ、TNF或细菌脂多糖(LPS)诱导的急性炎症持续24小时,未诱导WSTF丢失(图3a,b)。相比之下,低剂量、重复转染dsDNA或dsRNA在8天后降低WSTF蛋白水平,且未诱导衰老(通过p16和lamin B1检测)(图3c)。WSTF的丢失分别由cGAS响应胞质dsDNA或MAVS响应胞质dsRNA介导(扩展数据图9c,d)。慢性IFNβ处理也观察到WSTF丢失(图3d)。
作者进一步发现,慢性炎症中WSTF的丢失由自噬驱动。WSTF在慢性而非急性条件下发生核-胞质转运,并被自噬体和溶酶体靶向(图3e,f)。自噬基因ATG7或FIP200的破坏阻碍了慢性条件下WSTF的丢失(图3g)。一致地,GABARAP-WSTF结合在慢性而非急性核酸转染中增加。与衰老类似,ATM参与WSTF下调,ATM抑制剂ATMi处理阻碍了慢性炎症条件下WSTF的丢失。慢性而非急性核酸转染的细胞中观察到ATM激活,与许多慢性炎症条件促进DNA损伤反应的观点一致。这些结果表明,WSTF核自噬特异性发生于慢性而非急性炎症条件下。
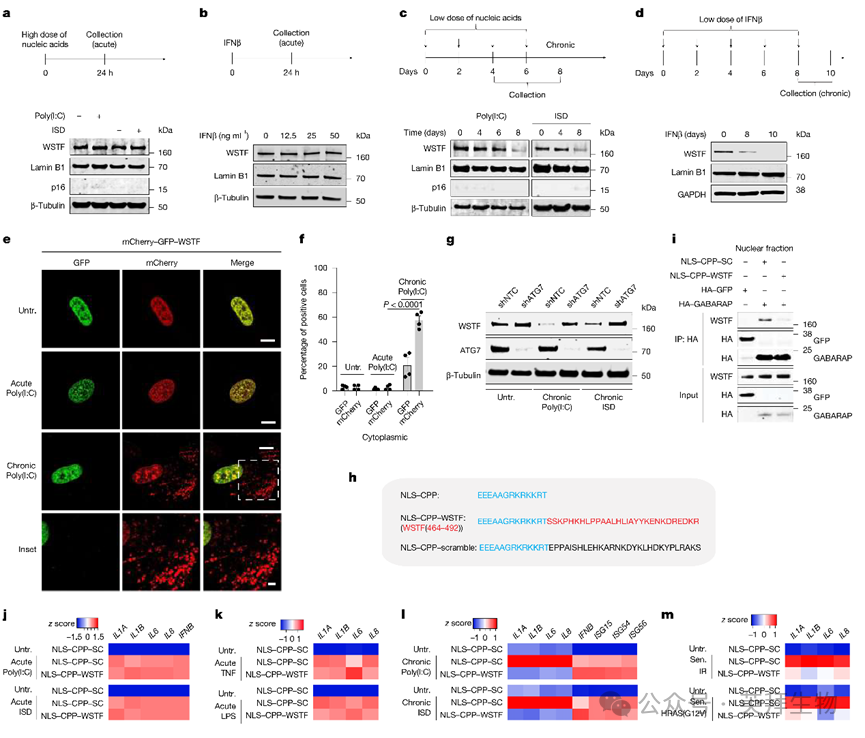
图3:WSTF在急慢性炎症中的作用
8.设计稳定WSTF的肽
作者研究了恢复WSTF蛋白作为抑制慢性炎症的治疗方法的潜力,重点阻断WSTF自噬降解所需的GABARAP-WSTF相互作用。源自WSTF 466-492区域的肽与全长WSTF竞争结合GABARAP。WSTF的计算机建模显示其GABARAP结合区域形成α-螺旋,并进一步提供了WSTF优先结合GABARAP而非LC3B的结构见解。
作者将WSTF 464-492区域的氨基酸序列与细胞穿透核定位信号(NLS)肽(NLS-CPP)融合,后者源自Glu-OCT6,包含OCT6的NLS,以促进核递送33。这生成了NLS-CPP-WSTF及其乱序对照(图3h)。加入细胞培养基后,NLS-CPP-WSTF在全细胞提取物和核组分(图3i)中抑制WSTF-GABARAP结合。
作者在急性和慢性炎症条件下测试了WSTF细胞穿透肽。加入培养基的NLS-CPP-WSTF在急性炎症中对炎症基因表达影响甚微(图3j,k)。相比之下,在双链核酸转染诱导的慢性炎症中,NLS-CPP-WSTF保留了WSTF蛋白水平并减少炎症细胞因子表达(图3l)。恢复WSTF抑制慢性炎症中的炎症细胞因子表达,但不影响限制病原体感染所需的IFNβ或干扰素刺激基因(图3l)。此外,NLS-CPP-WSTF减弱了衰老人细胞中WSTF的下调和炎症基因表达(图3m)。为扩展这些发现,作者基于小鼠WSTF序列(与人源序列相差4个残基)设计了小鼠肽mNLS-CPP-WSTF及其乱序对照mNLS-CPP-SC。加入培养基的mNLS-CPP-WSTF抑制了衰老小鼠细胞中WSTF的下调和炎症基因表达。
因此,抑制GABARAP-WSTF相互作用抑制慢性炎症而非急性条件下的炎症细胞因子表达。此外,在慢性条件下恢复WSTF蛋白水平不影响限制病原体感染所需的干扰素反应。WSTF的这些独特特征提示了干预慢性疾病的治疗机会,作者将在小鼠模型和人类患者样本中展示。
9. 在MASH中靶向WSTF
MASH是一种以慢性炎症、纤维化和肝损伤为特征的进展性肝病。MASH的治疗选择有限。作者评估了人类患者的肝样本,发现与无MASH的对照个体相比,MASH患者核内WSTF显著减少,在两个不同的MASH患者队列中进行了评估(图4a,b)。
作者使用缺乏蛋氨酸胆碱、补充乙硫氨酸(MCDE)饮食的小鼠MASH模型36,37,与正常饮食的小鼠进行比较。MCDE饮食小鼠的肝脏WSTF蛋白水平低于对照组。MCDE条件下WSTF可在胞质中检测到,部分与LAMP1共定位。小鼠在MCDE处理期间每日腹腔注射mNLS-CPP-WSTF肽或mNLS-CPP-SC肽。mNLS-CPP-SC肽注射的小鼠肝脏WSTF蛋白显著丢失,而mNLS-CPP-WSTF肽注射的小鼠肝脏保留了WSTF蛋白。作者进一步评估了这些小鼠肝脏的炎症,发现mNLS-CPP-WSTF肽处理的小鼠巨噬细胞(由F4/80标记)积累减少,炎症细胞因子表达受损。此外,mNLS-CPP-WSTF肽处理的小鼠肝纤维化减轻,通过α-SMA和Sirius Red染色测量。
除肽注射外,作者使用由甲状腺素结合球蛋白(TBG)启动子驱动的AAV2/8载体,实现小鼠WSTF 407-553区域的肝特异性表达。该短WSTF片段(人或小鼠源)与3NLS融合,结合GABARAP并抑制GABARAP-WSTF相互作用,从而抑制慢性炎症条件下WSTF降解。作者通过眶后注射给小鼠施用3NLS-mWSTF(407-553)-HA,使用3NLS-GFP-HA作为对照。
作者使用另一种饮食——缺乏胆碱、L-氨基酸定义的高脂饮食(CDAA-HFD)诱导MASH38。CDAA-HFD启动4周后,每月给小鼠施用AAV2/8载体一次;所有小鼠在12周后收集。3NLS-mWSTF(407-553)-HA表达抑制WSTF丢失(扩展数据图11h,i),减少巨噬细胞和纤维化标志物(扩展数据图11j,k),并抑制肝脏炎症基因表达(扩展数据图11l)。作者进一步使用Gubra-Amylin NASH(GAN)饮食诱导小鼠慢性MASH39(图4c)。GAN饮食启动3个月后,每月通过眶后注射给小鼠施用AAV2/8载体一次,所有小鼠在GAN饮食启动6个月后收集(图4d)。GAN饮食处理后,对照载体注射组WSTF减少,而3NLS-mWSTF(407-553)-HA组抑制了这种减少(图4e)。尽管GAN饮食增加了肝脏巨噬细胞和纤维化标志物,3NLS-mWSTF(407-553)-HA组的增加显著较低(图4f,g),与促炎基因表达上调减少一致(图4h)。
综上,这些数据表明WSTF在MASH肝脏中丢失,恢复WSTF蛋白抑制慢性炎症和纤维化(MASH的两个关键疾病特征)。未来对其他MASH临床方面的研究需进一步加强WSTF恢复的治疗潜力。
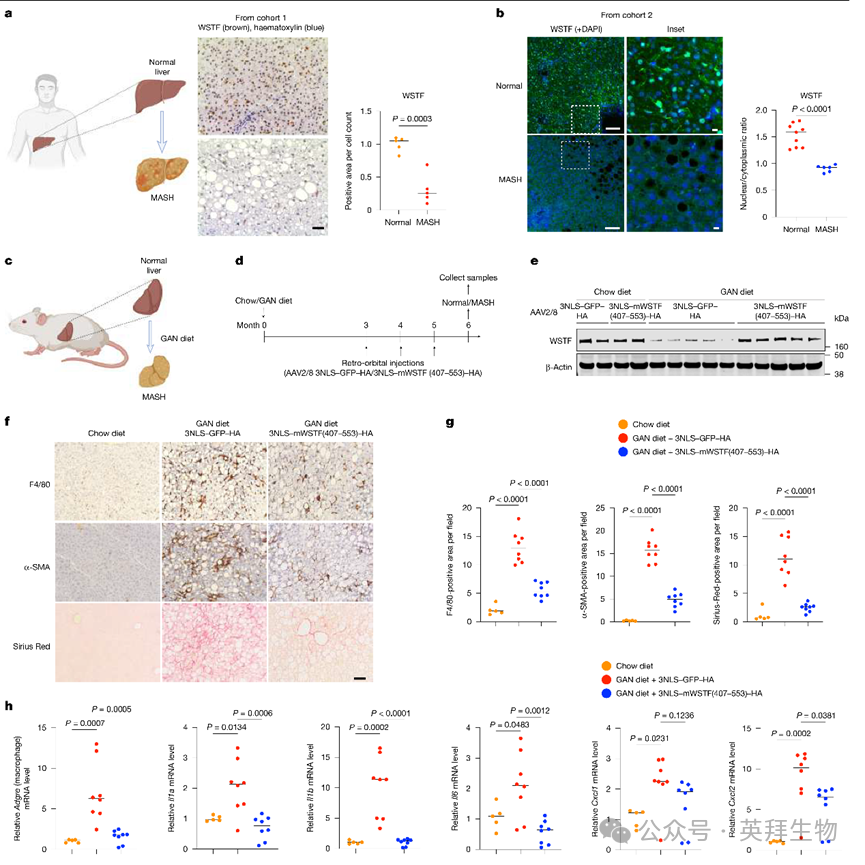
图4:靶向WSTF治疗MASH
10. 在OA中靶向WSTF
骨关节炎(OA)是另一种与慢性炎症相关的疾病,促进关节疼痛和软骨损伤40,41。为测试靶向WSTF在OA中的潜力,作者从接受膝关节置换的OA患者获取人关节软骨。收集相同区域等大小的关节软骨小块,在培养基中离体培养。这些外植体未处理或用NLS-CPP-SC或NLS-CPP-WSTF肽处理,随后分析条件培养基和外植体的炎症细胞因子产生(图5a)。如预期,不同组的外植体(如A、B、C;图5a)分泌不同水平的炎症细胞因子(图5b),由于关节软骨内炎症状态的异质性。然而,在所有三组中,NLS-CPP-WSTF肽处理的样本显示IL-6和IL-8分泌减少(图5b)。一致地,通过外植体切片的免疫组化(IHC)分析,NLS-CPP-WSTF肽减少了IL-6、IL-8和MMP13(OA中降解软骨基质的关键胶原酶40,41)。
为进一步减少OA软骨炎症严重程度的区域异质性,作者酶解患者样本股骨髁小块软骨以获得软骨细胞。NLS-CPP-WSTF肽处理一致抑制IL-6、IL-8和MMP13的表达水平。
作者接下来使用OA小鼠模型测试WSTF肽的治疗效能。手术切断内侧半月板韧带和前交叉韧带42,对照组小鼠进行假手术(不切断韧带)。该模型在2周内诱导膝关节关节损伤和慢性炎症。每隔一天关节内注射WSTF肽(图5c)。OA诱导手术导致WSTF从核转移至胞质,部分与LAMP1共定位,以及IL-8和MMP13显著增加。mNLS-CPP-WSTF肽减少了软骨中这两种细胞因子的诱导(图5d,e)。作者接下来评估关节损伤程度。每100 μm获取5 μm切片,用Safranin O染色,由三位独立观察者根据OARSI分级系统43盲法评分。评估显示mNLS-CPP-WSTF肽减轻软骨损伤(图5f,g),与炎症是关节炎软骨损伤驱动力44的观点一致。
总之,作者的结果表明NLS-CPP-WSTF肽在人类患者样本和小鼠中缓解OA后的慢性炎症。
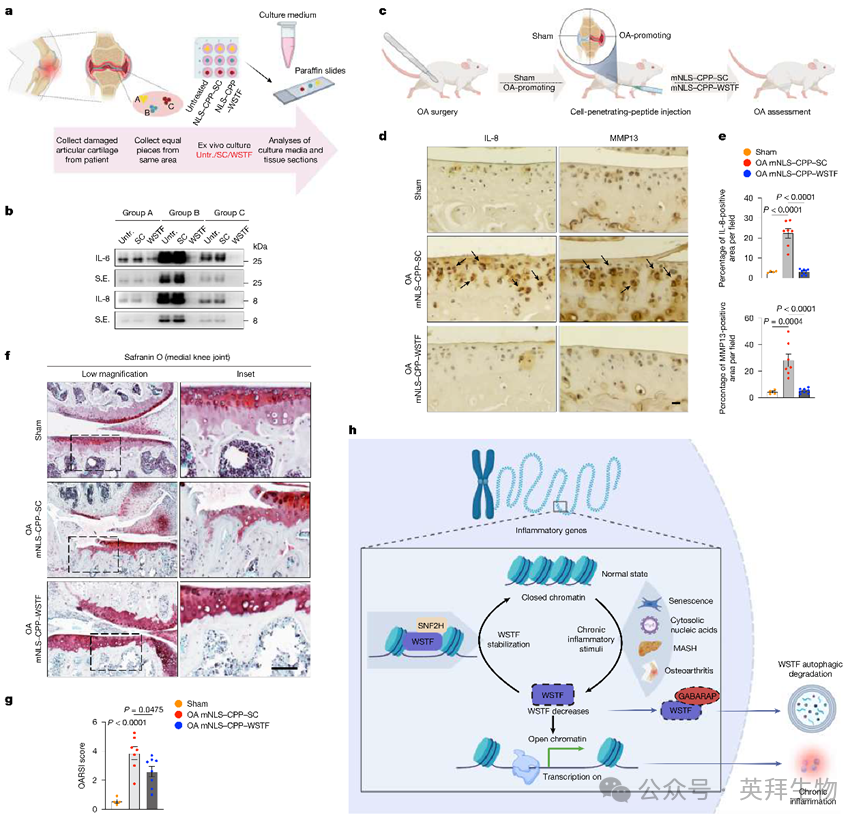
图5:靶向WSTF治疗骨关节炎
结论:
综上所述,本研究发现,核自噬通过ATM-GABARAP途径选择性降解染色质重塑蛋白WSTF,导致炎症基因染色质开放并放大慢性炎症,而这一机制在急性炎症中不发生;通过阻断WSTF与ATG8的相互作用恢复WSTF水平,可在小鼠模型及患者样本中特异性抑制MASH、骨关节炎等慢性炎症,同时保留急性免疫防御功能,为区分和治疗慢性炎症提供了新靶点与策略
参考文献:
Wang Y, Eapen VV, Liang Y, Kournoutis A, Sherman MS, Xu Y, Onorati A, Li X, Zhou X, Corey KE, Du K, Cabral Burkard AM, Ho CK, Xie J, Zhang H, Maeso-Díaz R, Ma X, Rieprecht U, O'Brien T, Cetinbas M, Wang L, Liu J, Bretz C, Havas AP, Zhou Z, Ho Sui SJ, Saladi SV, Sadreyev RI, Adams PD, Kingston RE, Diehl AM, Alman B, Goessling W, Yue Z, Wang XF, Johansen T, Dou Z. WSTF nuclear autophagy regulates chronic but not acute inflammation. Nature. 2025 Jul 2. doi: 10.1038/s41586-025-09234-1. Epub ahead of print. PMID: 40604282.