






吃对脂肪酸,抗癌更强大?油酸与棕榈酸对γδ-T细胞抗肿瘤免疫的影响,速来了解!
2型免疫反应是机体抵御寄生虫感染的重要防御机制,但过度的2型免疫反应会引发一系列病理性变应性疾病,如哮喘、食物过敏和特应性皮炎。该反应起始于上皮细胞释放的警报素(Alarmins),包括胸腺基质淋巴细胞生成素(TSLP)、白细胞介素-25(IL-25)和IL-33。这些因子触发级联反应,最终释放标志性的2型细胞因子IL-4、IL-5和IL-13(4)。这些细胞因子激活嗜酸性粒细胞、肥大细胞、第2组固有淋巴细胞(ILC2s)和辅助性T细胞2(TH2细胞),共同介导以粘液过度分泌、组织重塑和过敏性炎症为特征的2型免疫应答(5-7)。靶向IL-4/IL-13和TSLP的生物制剂在多种2型过敏性疾病中的临床应用及疗效,进一步证实了这些细胞因子的过度产生与疾病发生的内在关联。然而,调控2型免疫反应失调的内在细胞与分子机制,特别是其稳态抑制机制,尚未完全阐明。在此,研究人员揭示了肠道绒毛细胞,一种以触发2型免疫激活而闻名的特殊上皮细胞,还具有抑制2型反应的分子回路。在绒毛细胞中敲除转录因子Spi-B即足以引发自发性2型炎症。绒毛细胞内源性缺乏Spi-B使原本对食物过敏模型具有抵抗力的C57BL/6J小鼠变得易感。Spi-B抑制了由c-Kit信号驱动的绒毛细胞产生2型警报素TSLP。破坏这一负调控轴会导致绒毛细胞增生,并加剧2型炎症,这可以通过酪氨酸激酶抑制剂进行药物靶向治疗。这些发现确定了2型免疫反应中以绒毛细胞为中心的关键检查点,并突出了绒毛细胞在促进和抑制2型反应中的双重作用。本文于2025年7月发表于《Signal Transduction and Targeted Therapy》,IF 52.7。
技术路线
主要实验结果:
1. 转录因子Spi-B的缺失可诱发小肠簇细胞过度增生及自发性2型炎症
为探究小肠簇细胞的内在调控机制,我们对野生型(WT)小鼠经流式分选获得的上皮细胞黏附分子阳性(EpCAM+)SiglecF+簇细胞进行了RNA-seq分析。在簇细胞的转录因子编码基因中,编码Ets家族转录因子Spi-B的Spib基因呈现为簇细胞内表达水平最高且最具特异性的转录本之一(图1A)。对已发表的小肠上皮细胞(IECs)scRNA-seq数据的分析进一步证实:Spib在簇细胞群体中呈现优势性高表达,其表达水平甚至超过公认的簇细胞主调控因子基因Pou2f3(图1B)。重新分析已公开的RNA-seq数据显示,Spib在多种小鼠组织的簇细胞中均保持持续高表达。为验证上述发现,我们通过小鼠小肠组织的免疫荧光染色原位检测Spi-B表达。共聚焦显微镜分析表明,无论是否感染蠕虫Heligmosomoides polygyrus,Spi-B均与簇细胞标志物双皮质素样激酶1(DCLK1)存在共定位(图1C)。综上,这些发现凸显了Spi-B作为一种高度特异性于杯状细胞的标志性转录因子的重要性。
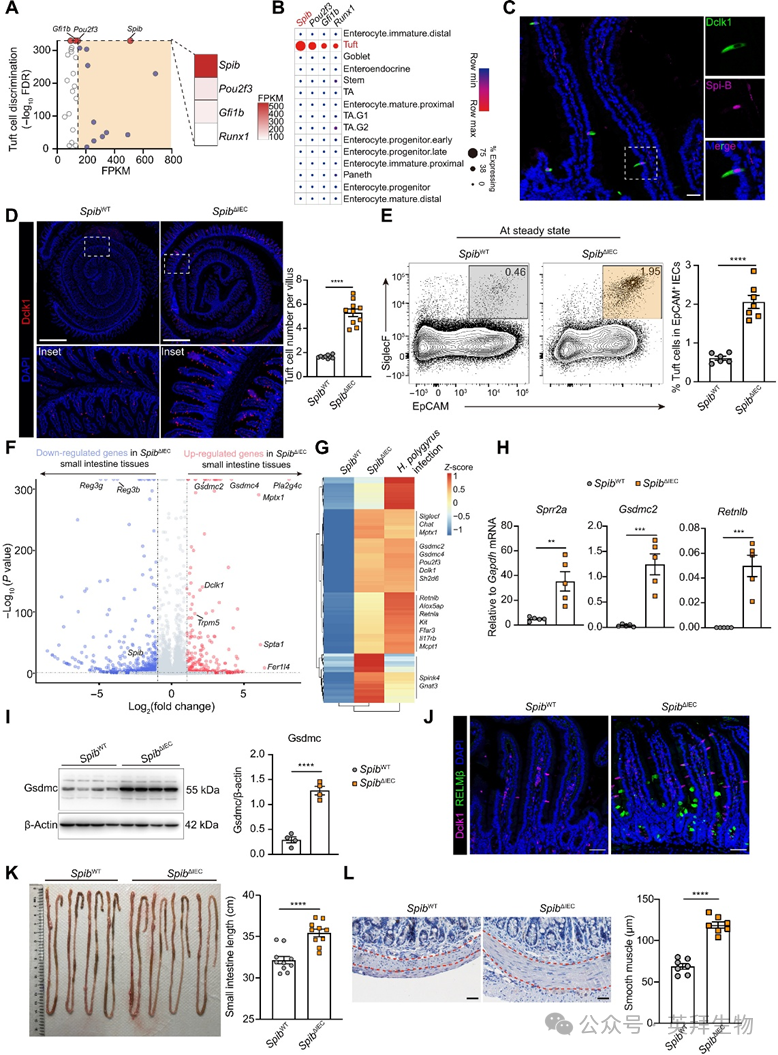
图1.上皮Spi-B缺失会导致肠道黏膜自发性出现2型炎症
为阐明Spi-B在簇细胞中的作用,我们构建了Spibfl/flVillin-Cre小鼠(下文简称SpibΔIEC),实现肠道上皮细胞(IECs)特异性Spib基因缺失。Spi-B缺失导致簇细胞数量与比例显著增加,稳态条件下较同窝对照小鼠升高3-6倍(图1D-E)。同时,Spib缺陷小鼠的杯状细胞数量及黏液分泌同步增加,但肠道绒毛整体结构及其他上皮细胞亚群(如潘氏细胞、增殖细胞)基本无变化。5-溴-2′-脱氧尿苷(BrdU)示踪实验证实Spib缺失不影响IECs整体增殖。此外,扫描电镜分析未检测到三毛滴虫定植迹象,表明表型与原生动物感染无关。
为了全面评估Spi-B缺失所导致的转录变化,我们对来自未感染的SpibΔIEC小鼠和对照小鼠的小肠组织进行大规模RNA-seq分析发现:SpibΔIEC小鼠中簇细胞特异性基因(如Dclk1、Trpm5)及蠕虫感染相关转录本显著上调(图1F)。抗蠕虫基因(如Pla2g4c、Gsdmc2)的上调促使我们比较SpibΔIEC小鼠与感染Heligmosomoides polygyrus蠕虫14天(dpi)的野生型(WT)小鼠表达谱。Spib缺陷小鼠中大多数上调基因呈现与蠕虫感染诱导基因高度相似的表达模式(图1G)。基因集富集分析(GSEA)进一步显示,Spib缺陷小鼠中tuft-1与tuft-2细胞群均显著富集,其中tuft-2细胞富集程度更高。以上表明:Spib缺失可诱导抗蠕虫基因程序表达,形成“类蠕虫感染”转录谱,即使在没有蠕虫的情况下也是如此。
为验证Spi-B缺失是否导致功能表型模拟,我们检测蠕虫感染效应分子表达:Spib缺陷小鼠IECs中Sprr2a、Gsdmc2和Retnlb显著上调(图1H),这些基因在蠕虫感染中强诱导。其中Gasdermin C(Gsdmc)与抵抗素样分子β(RELMβ)在蠕虫感染中高表达,分别介导IL-33释放和蠕虫排出。与转录水平一致,SpibΔIEC小鼠小肠Gsdmc与RELMβ蛋白表达同步升高(图1I-J),提示出现了活跃的2型免疫应答。除簇细胞扩增和上皮重塑外,SpibΔIEC小鼠还表现出小肠长度增加与肌层增厚(图1K-L),此表型与蠕虫感染后簇细胞增生特征一致。综上,Spi-B缺失可诱发自发性2型炎症状态,其特征为簇细胞增生、抗蠕虫效应分子上调和类蠕虫感染的肠道重塑,证实Spi-B在抑制过度2型炎症中的关键作用。
2. 簇细胞固有的Spi-B可抑制蠕虫及食物过敏原诱导的2型应答
为明确Spi-B在簇细胞中的固有功能,我们构建并验证了他莫昔芬诱导型Spibfl/flPou2f3-CreERT2小鼠(图2A-B,下文简称SpibΔTuft)。尽管SpibΔIEC小鼠出现预期微皱褶细胞(M细胞)缺失,SpibΔTuft小鼠仍维持正常M细胞发育,证实簇细胞特异性Spib缺失。与SpibΔIEC表型一致,簇细胞特异性Spi-B缺失在稳态下导致簇细胞增生(经Dclk1与绿色荧光蛋白(GFP)标记Pou2f3共染色证实)(图2C-D)。伴随簇细胞数量/频率增加,SpibΔTuft小鼠较同窝对照呈现小肠延长与肌层增厚(图2E-G)。上述表型重现证实:Spi-B以簇细胞固有方式限制其过度扩增并维持组织稳态。
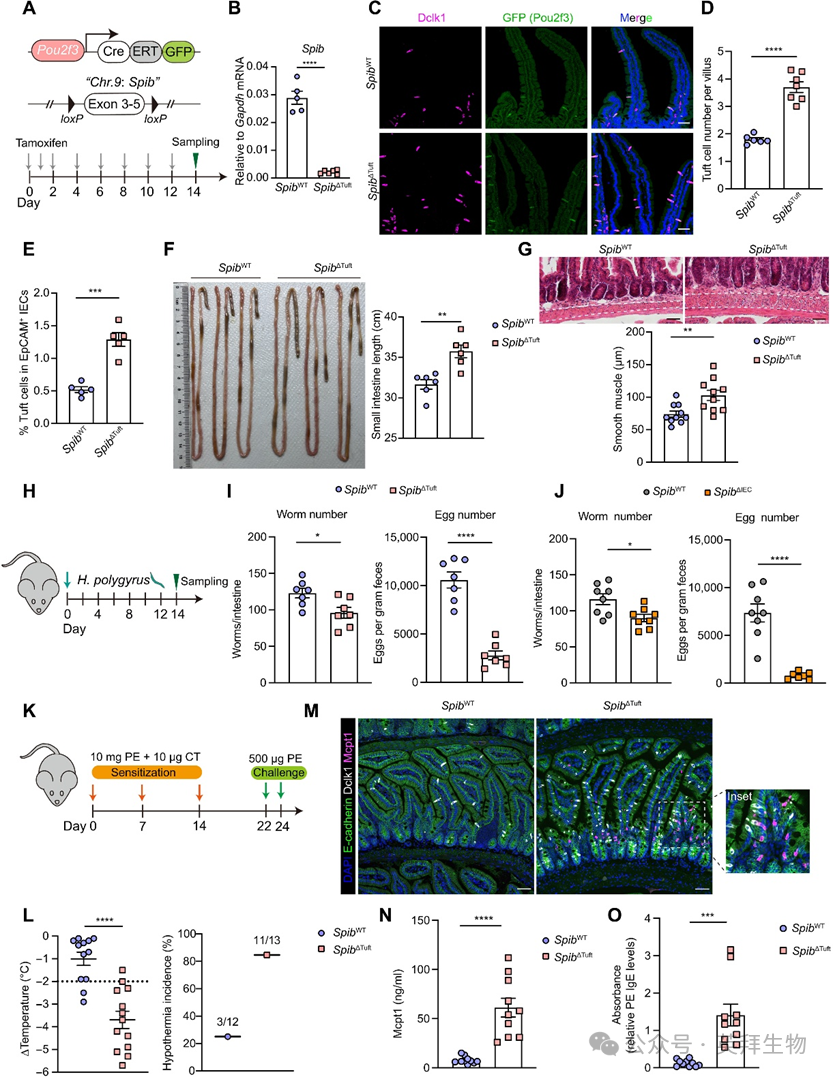
图2. 簇细胞固有Spi-B抑制簇细胞过度扩增并调控抗蠕虫感染与食物过敏原的2型免疫应答
为验证稳态下观察到的差异是否影响宿主对2型刺激的应答,我们对SpibΔTuft小鼠及对照进行Heligmosomoides polygyrus幼虫感染(图2H)。与Spi-B充足对照组相比,SpibΔTuft小鼠表现出虫荷与虫卵负荷同步下降,提示抗蠕虫感染能力增强(图2I)。一致地,SpibΔIEC小鼠也显示更强的蠕虫防御能力,具体表现为虫荷减少、产卵量下降、杯状细胞数量增加及黏液分泌增多(图2J)。
除抗蠕虫免疫外,2型免疫应答在影响全球10%人群的食物过敏发病机制中起核心作用。为评估Spi-B缺失对食物过敏原易感性的影响,我们采用花生提取物(PE)建立食物过敏模型(图2K)。在通常具有抵抗力的C57BL/6J背景下,SpibΔTuft小鼠表现出核心体温下降及低体温发生率上升,表明对食物过敏原致敏的敏感性增强(图2L)。膳食抗原引发的过敏反应源于肥大细胞扩增与活化,其标志是肥大细胞蛋白酶-1(Mcpt1)水平升高。经PE再次激发后,SpibΔTuft小鼠较对照组呈现:上皮内Mcpt1+肥大细胞数量增加、血清Mcpt1浓度升高以及花生特异性免疫球蛋白E(IgE)水平上升(图2M-O)。过敏原激发后还观察到肥大细胞与嗜酸性粒细胞比例增加。综上表明:簇细胞固有Spi-B通过抑制蠕虫感染与食物过敏原引发的过度2型免疫应答,维持免疫稳态。
3. 簇细胞固有Spi-B通过调控TH2细胞应答抑制2型免疫
为阐明Spi-B缺失驱动自发性2型免疫的机制,我们在小肠类器官中发现:无论是否存在IL-4,Spi-B缺失均不影响簇细胞分化,表明Spi-B不直接调控肠干细胞向簇细胞的谱系定向。随后通过固有层(LP)免疫细胞区室免疫表型分析探究簇-免疫细胞互作。结果显示:嗜酸性粒细胞、巨噬细胞及树突细胞等先天免疫细胞群比例无显著变化;SpibΔIEC小鼠与对照组的ILC2s比例及其IL-13分泌水平亦相当,提示ILC2s在Spib缺失背景下贡献有限。
在小肠固有层CD4+ T辅助细胞亚群中,SpibΔIEC小鼠呈现明显的TH2表型极化(图3A-B)。与对照相比,其GATA3+ TH2细胞比例与数量增加,而RORγt+ TH17细胞频率降低(图3B-C)。细胞因子检测进一步显示:Spib缺陷小鼠TH2细胞的IL-13分泌增加,但IL-4分泌未变(图3D)。在SpibΔTuft小鼠中也观察到TH2细胞丰度与IL-13分泌增强(图3E-F),证实Spi-B以簇细胞固有方式调控TH2应答。信号转导与转录激活因子6(STAT6)作为IL-13信号激活的关键转录因子,可驱动肠上皮簇细胞增生。与TH2细胞IL-13分泌增加一致,免疫染色显示SpibΔIEC小鼠上皮细胞内STAT6酪氨酸磷酸化水平升高(图3G-H)。
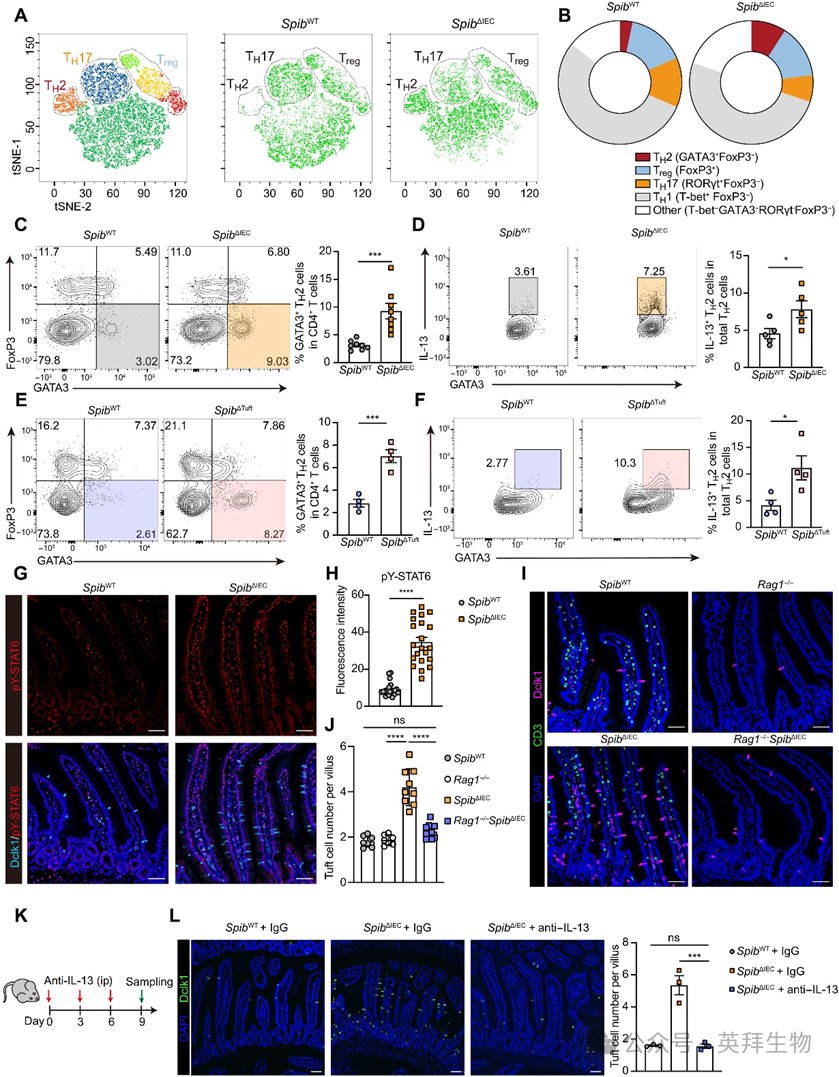
图3. Spi-B通过调控TH2细胞应答抑制肠道簇细胞扩增
为阐明适应性免疫细胞的作用,我们构建了Rag1−/− SpibΔIEC小鼠。Rag1缺失可消除SpibΔIEC小鼠的自发性2型炎症表型,包括簇细胞增生、上皮重塑及平滑肌增厚(图3I-J)。此外,IL-13中和抗体处理能消除SpibΔIEC小鼠的簇细胞增生(图3K-L),表明过量IL-13是Spi-B缺失诱导簇细胞增生的关键驱动因子。综上证明:Spi-B通过限制TH2细胞丰度及反应性,内在调控簇细胞扩增。
4. 簇细胞源性TSLP驱动Spi-B缺失诱导的簇细胞增生
为探究Spi-B调控TH2活性及簇细胞增生的内在机制,我们对分选的野生型(SpibWT)与SpibΔIEC小鼠簇细胞进行批量RNA测序(图4A)。警报素细胞因子基因Tslp在Spib缺陷簇细胞中上调最显著(图4A)。多组独立实验证实:SpibΔIEC与SpibΔTuft小鼠簇细胞的Tslp mRNA表达量较对照组升高6-8倍,而另一警报素Il25表达不变(图4B-C)。小肠分泌性TSLP蛋白水平在Spib缺陷小鼠中同步升高(图4D-E)。单分子荧光原位杂交(smFISH)显示Tslp转录本特异性富集于Spib缺陷簇细胞,证实其为TSLP升高的主要来源(图4F-G)。
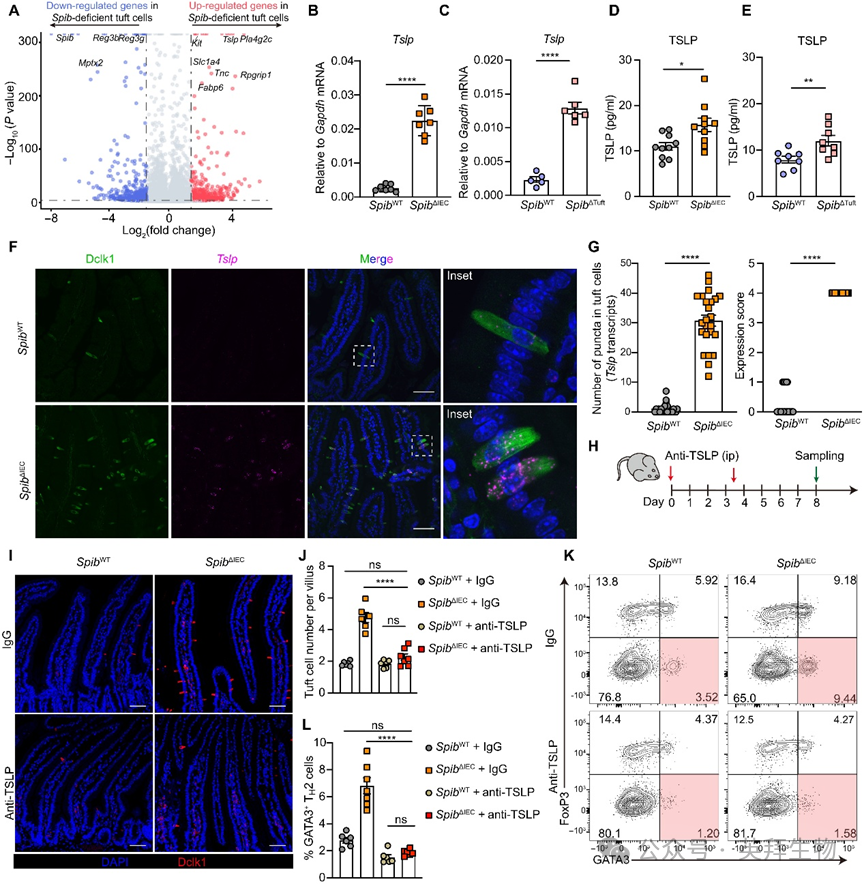
图4. 簇细胞源性TSLP参与Spi-B缺失触发的簇细胞扩增
鉴于TSLP的强效2型免疫诱导能力,我们进一步验证其功能贡献:给予TSLP中和抗体可消除Spib缺陷小鼠的簇细胞数量增加、TH2细胞比例升高、STAT6活化及RELMβ产生(图4H-L),证实TSLP在该自发性2型应答中不可或缺。虽然ILC2s高表达Il17rb,但TH2细胞呈现更高TSLP受体(TSLPR,由Crlf2编码)水平。为探究CD4+ T细胞TSLPR在簇细胞增生中的作用,我们将CRISPR-Cas9体外编辑的Crlf2缺陷型或对照CD4+ T细胞过继移植至Rag1−/−SpibΔIEC小鼠(图5A)。尽管固有层CD4+ T细胞比例相似,但接受sgCrlf2 CD4+ T细胞的小鼠表现出簇细胞数量减少、TH2细胞频率降低及Retnlb表达下降(图5B-C),证明CD4+ T细胞的TSLPR促进Spi-B缺失诱导的2型炎症。此外,广谱抗生素处理可消除Spib缺陷小鼠的簇细胞增生,提示微生物群在此条件下参与簇细胞扩增。
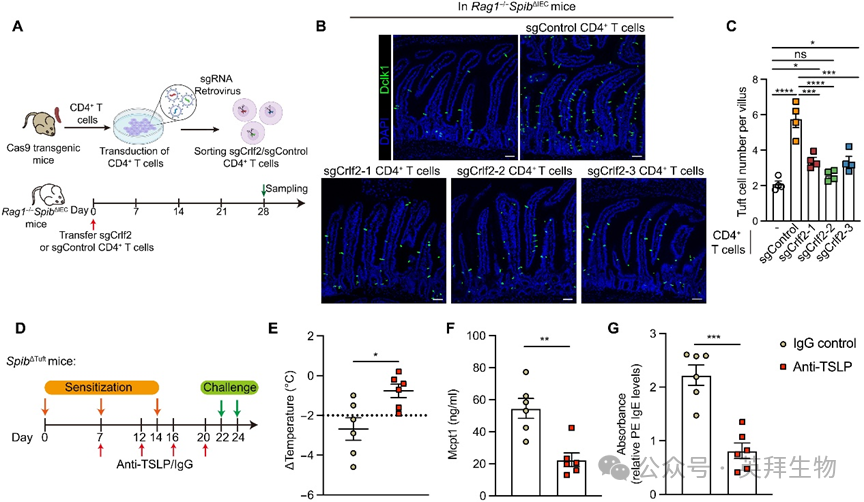
图5. 簇细胞源性TSLP促进Spi-B缺陷小鼠的CD4+ T细胞依赖性簇细胞增生及过敏应答
为验证TSLP升高是否介导SpibΔTuft小鼠的食物过敏症状,TSLP阻断处理有效阻止了特征性体温下降并降低血清Mcpt1与IgE水平(图5D-G)。综上表明:Spi-B在稳态下抑制簇细胞TSLP产生,其缺失导致的簇细胞源性TSLP升高驱动簇细胞增生。
5. 簇细胞Spi-B对Kit的转录抑制
为解析TSLP抑制机制,我们采用靶向切割与转座酶技术(CUT&Tag)分析簇细胞Spi-B-DNA互作(图6A)。Spi-B结合峰广泛分布于基因组各区域(图6B)。整合Spi-B CUT&Tag启动子结合位点与RNA-seq上调基因(图4A),鉴定出22个重叠基因(图6C)。其中Kit在Spib缺陷簇细胞中上调最显著。CUT&Tag显示:Spi-B在野生型簇细胞中直接结合Kit启动子,该结合在缺陷细胞中消失(图6D);而Tslp、Il25等基因调控区无特异性结合。SpibΔIEC与SpibΔTuft小鼠中均证实Kit/c-Kit在mRNA与蛋白水平表达升高,且c-Kit高表达簇细胞比例增加(图6E-H)。为明确c-Kit升高是否为上皮细胞自主现象,小肠类器官实验显示:无免疫细胞存在时,Spib缺陷簇细胞仍呈现c-Kit上调(图6I)。荧光素酶报告基因实验证实Spi-B以剂量依赖方式转录抑制Kit启动子,且该抑制依赖其完整的反式激活与DNA结合域(图6J)。
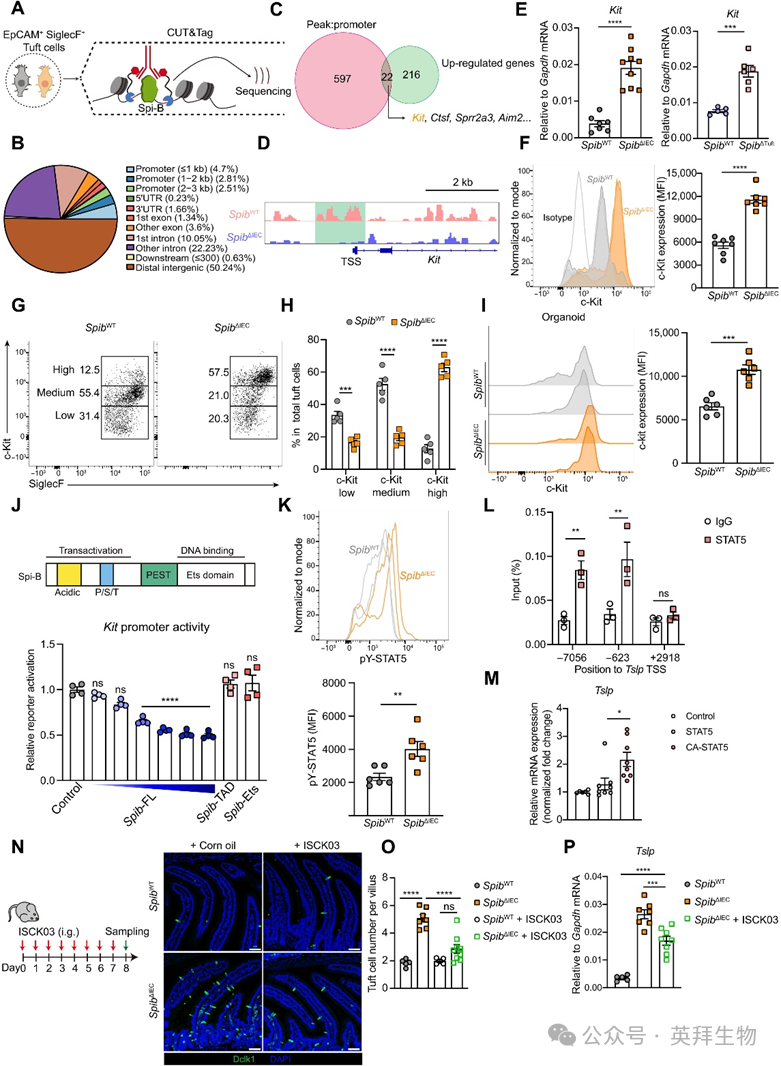
图6. Spi-B在簇细胞内源性抑制Kit表达
伴随c-Kit表达增强,Spib缺陷簇细胞中STAT5磷酸化水平(c-Kit信号标志)升高(图6K)。STAT5在小肠上皮细胞中结合内源性Tslp基因座的预测增强子与启动子区(非基因体区)(图6L)。在CMT-93上皮细胞中表达组成型活化STAT5(CA-STAT5)可增加Tslp转录(图6M),提示c-Kit-STAT5信号转录驱动TSLP表达。为验证c-Kit信号对TSLP诱导及簇细胞增生的必要性,采用c-Kit抑制剂ISCK03(强效选择性抑制剂)进行药理学干预,可有效缓解Spib缺陷小鼠的簇细胞增生、Tslp表达、TH2细胞比例及IL-13分泌(图6N-P)。蠕虫感染与食物过敏模型中,簇细胞呈现Spib下调伴随c-Kit与Tslp升高,表明该通路在此类条件下被激活。综上揭示:Spi-B作为Kit转录抑制因子,通过限制c-Kit表达抑制TSLP产生。
6. 簇细胞固有Spi-B-Kit-TSLP回路调控肠道2型免疫
为明确c-Kit在簇细胞中的功能,我们构建并验证Kitfl/flPou2f3-CreERT2(KitΔTuft)小鼠,进一步与Spibfl/fl杂交获得簇细胞特异性双敲除(SpibΔTuftKitΔTuft)小鼠。他莫昔芬诱导基因缺失后,双敲除小鼠较SpibΔTuft小鼠表现出绒毛内簇细胞数量与频率降低(图7A-C)。Kit缺失导致类器官簇细胞STAT5磷酸化减弱,体内外Tslp表达下降及TH2细胞比例减少(图7D-E),证实c-Kit是Tslp表达与TH2介导2型免疫病理的上游调控因子。表型上,簇细胞c-Kit缺失削弱SpibΔTuft小鼠的抗蠕虫防御增强效应,表现为虫卵负荷增加与上皮Retnlb表达降低(图7F)。同时c-Kit缺失改善食物过敏表型:核心体温回升、血清Mcpt1下降及肥大细胞扩增减弱(图7G-H)。上述发现共同证明:簇细胞固有Spi-B-Kit-TSLP轴对维持簇细胞稳态及应答激活刺激后的2型免疫至关重要。
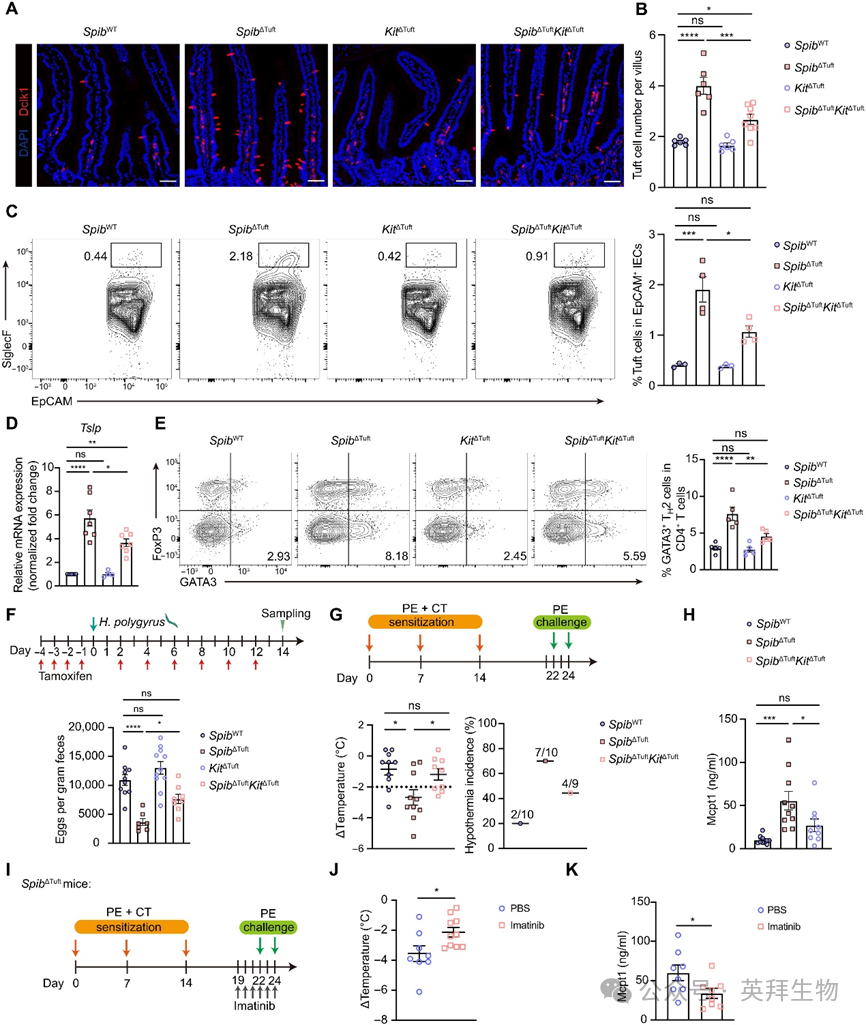
图7. 簇细胞固有Spi-B通过限制c-Kit-TSLP轴抑制2型免疫
为探索靶向c-Kit过度活化在食物过敏中的治疗潜力,我们使用FDA批准酪氨酸激酶抑制剂伊马替尼(Gleevec)(图7I)。与机制研究选用ISCK03不同(图6N),伊马替尼因其成熟的临床应用及治疗c-Kit依赖性疾病(如慢性髓系白血病)的安全性被选为治疗模型。伊马替尼处理显著减轻SpibΔTuft与野生型小鼠的食物过敏反应发展与严重程度,表现为体温恢复及血清Mcpt1降低(图7J-K)。靶向c-Kit通路是潜在的过敏治疗策略。
Spi-B缺失背景下,TSLP上调与2型应答过度活化至少部分由c-Kit信号介导。c-Kit作为受体酪氨酸激酶,在造血干细胞与肥大细胞等增殖分化中起关键作用。肠道中c-Kit功能见于肠干细胞、潘氏细胞与杯状细胞,但其在簇细胞中的作用未知。可能因野生型簇细胞c-Kit基础表达极低导致研究缺失。进一步遗传学剔除该低水平c-Kit未引起簇细胞丰度与功能的显著改变(图7C-D)。簇细胞低c-Kit表达非被动事件,而是Spi-B主动转录抑制的结果。该抑制解除后,c-Kit高表达驱动TSLP介导的簇细胞增生,甚至使C57BL/6J小鼠对食入性过敏原易感。食物过敏中簇细胞Spib下调伴随c-Kit与Tslp升高支持该通路的临床相关性。使用临床药物伊马替尼抑制c-Kit过度活化可缓解过敏病理发展。但鉴于c-Kit在肥大细胞等广泛表达,且伊马替尼对其他酪氨酸激酶有抑制效应,c-Kit在簇细胞中的精确作用及治疗潜力需进一步研究。本研究为靶向簇细胞区室、调控Spi-B-Kit-TSLP轴以恢复免疫稳态和改善2型病理提供了机制框架。
参考文献:
Wang J, Shen R, Yang K, Guo X, Yu J, Wu C, Guo XK, Hu X. Tuft cells restrain intestinal type 2 immunity through the transcription factor Spi-B. Sci Immunol. 2025 Jul 4;10(109):eads5818. doi: 10.1126/sciimmunol.ads5818. Epub 2025 Jul 4. PMID: 40614214.