






不同的启动阶段通过协调旁分泌型 IL-2 信号调节 CD8 T 细胞免疫力
从T细胞的克隆增殖是适应性免疫的基石,这一过程的启动称为T细胞“启动”,依赖于T细胞与抗原呈递树突状细胞(DCs)的相互作用,涉及T细胞受体(TCR)激活、共刺激和炎症细胞因子。这一过程在次级淋巴器官中发生,持续约24小时。之后,T细胞脱离并与DCs分离,开始活跃迁移,并迅速增殖分化为效应T细胞和记忆T细胞。这篇文章的核心内容是关于CD8 T细胞在免疫反应中的激活和分化过程,特别是研究了在T细胞启动阶段,通过协调旁分泌型IL-2信号来调节CD8 T细胞免疫力的机制。作者通过实验模型和活体成像技术,揭示了CD8 T细胞在淋巴结中的动态行为以及与DCs和CD4 T细胞的相互作用,提出了CD8 T细胞启动的两个阶段模型,并强调了这些发现对疫苗接种和细胞免疫疗法的潜在影响。该研究于2024年10月发表在《Science》,IF 44.7分。
技术路线:
主要研究结果:
1 在时间上分离的启动阶段中的CD8 T细胞动态
首先,作者研究了在病毒感染后,病毒抗原在引流淋巴结中由DCs在何种微环境中呈现。作者进行了表达淋巴细胞性脉络丛脑膜炎病毒(LCMV)糖蛋白(GP)的天花病毒(VV-GP)的皮下感染,并转移了能够通过表达荧光tdTomato进行追踪的特异性TCR转基因CD8 T细胞(P14)。在感染后24小时内,P14 T细胞聚集在引流的腘淋巴结的滤泡间和皮质边缘区域(图1A),揭示了抗病毒CD8 T细胞的初始启动,其特征是与感染的DCs进行长期相互作用。在感染后第3天,这些T细胞已经增殖,并且近似均匀分布在淋巴结的副皮质中(图1A)。
为了测试在这一较晚时间框架内,呈现GP33的DCs是否存在于该区域,作者在感染后第3天转移了一组新的幼稚P14 T细胞。在8小时后,转移的T细胞上调了CD69并形成了簇,从而表明存在呈现GP33的DCs。对T细胞聚集的定位和丰度进行系统分析,通过主要组织相容性复合体I(MHC-I)识别病毒抗原的DCs,证实了在d3 p.i.时,抗原呈递发生在亚滤泡区域,并在d4 p.i.时显著下降(图1,B和C)。
由于其上方的大B细胞滤泡,亚滤泡区域不易通过IVM进入。然而,在一些罕见的情况下,使用报告小鼠(XCR1WT/Venus),作者观察到抗原特异性CD8 T细胞与亚滤泡区域的常规1型树突状细胞(cDC1s)之间数小时的相互作用。为了更深入地研究这些细胞簇,并可靠地通过IVM进入亚滤泡区域,作者使用了CD20单克隆抗体来耗尽覆盖这些区域的B细胞。证实B细胞耗竭并未影响病毒特异性细胞毒性T淋巴细胞的增殖和分化。在B细胞耗竭后,作者转移了幼稚的P14 T细胞,应用了CD62L阻断抗体,随后用VV-GP感染小鼠。在感染后第3天的引流腘淋巴结中,作者观察到大型P14 T细胞,很可能是被激活的T细胞母细胞(淋巴母细胞),停止迁移并与cDC1s相互作用至少1小时(图1,D到G)。
这表明,在初始启动后与DCs分离后迁移的CD8 T细胞在这些亚滤泡区域与抗原呈递的DCs重新建立了长期相互作用。作者假设,在亚滤泡淋巴结区域中T细胞母细胞与cDC1s之间的这些二次紧密细胞相互作用可能有潜力塑造最终的原发性CD8 T细胞反应:作者将此称为“第二次启动阶段”。
值得注意的是,亚滤泡淋巴结区域内的T细胞母细胞与多个局部聚集的cDC1s相互作用,并且可能还与作者的报告小鼠未突出显示的其他DCs相互作用(图1D)。这与病毒感染期间的初始启动形成对比,初始启动时多个幼稚CD8 T细胞通常在IFA中围绕单个DC形成簇。相应地,与在d3与DC网络相互作用时相比,T细胞在d1与单个DC相互作用时,簇内T细胞密度更高。由于d3时簇内CD8 T细胞的大小增加,作者询问被激活的T细胞是否继续或只是刚刚开始它们的增殖周期。为探究被激活的T细胞是否处于起始增殖周期,作者用增殖染料CMFDA(5-氯甲基荧光素二乙酸酯)标记幼稚的P14 T细胞和多克隆对照CD8 T细胞,将它们共同转移到野生型(WT)受体中,并在d3用IVM进行分析。在非特异性对照CD8 T细胞显示出明亮的CMFDA信号,而在停滞的P14 T细胞中,该信号已稀释至无法检测的水平(图1,H和I)。因此,与DCs重新相互作用的CD8 T细胞至少已经增殖过一次,导致CMFDA的荧光强度稀释。
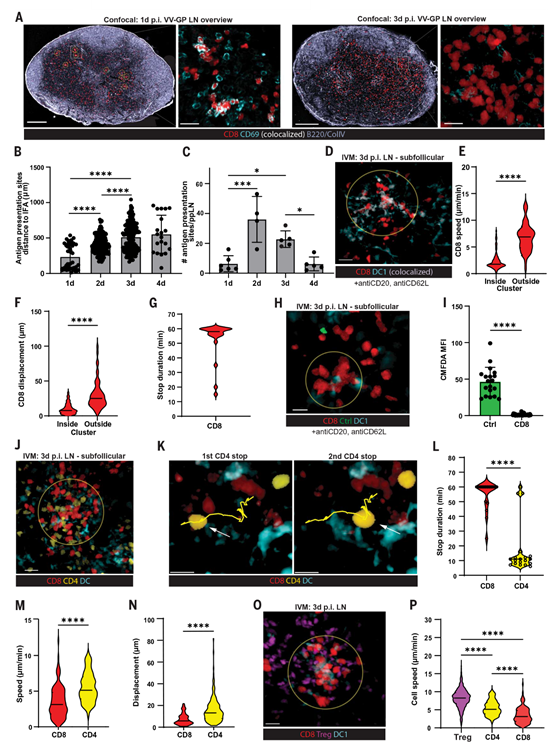
图1:在时间上分离的CD8 T细胞启动阶段的细胞动态
2 CD4 T细胞在时间上分离的启动阶段的动态
接下来,为了探究第二次启动阶段中,抗原特异性CD4 T细胞的定位情况,以及它们是否会像CD8 T细胞那样重新参与长期相互作用。作者将P14 CD8(tdTomato)和特异性GP的SMARTA CD4 T细胞(Venus)共同转移到CD11c-mCherry小鼠中,用VV-GP感染它们,并在用于检查CD8 T细胞的实验条件下,在第2天至第3天之间进行分析。作者发现SMARTA淋巴母细胞与停滞的P14 T细胞处于相同的生态位中(图1J)。值得注意的是,在这一启动阶段,SMARTA T细胞的迁移模式与P14 T细胞不同。SMARTA T细胞短暂停止迁移,平均与DCs相互作用10分钟(图1,K和L)。然后,它们以大约5 mm/min的较低速度在簇内的DCs之间移动(图1M)。与这种走走停停的迁移模式一致,SMARTA T细胞的细胞位移比与DCs通常在整个60分钟记录期间进行局部相互作用的P14 T细胞更高(图1N)。在一些罕见的情况下,SMARTA T细胞与DCs进行了至少持续60分钟的长期相互作用(图1L)。
已知CD4 Treg细胞可以控制抗病毒CD8 T细胞反应。为了可视化内源性、多克隆Treg细胞群的细胞动态,作者将Foxp3Cre ERT2-R26-tdTomato小鼠(通过他莫昔芬处理可以选择性地在Foxp3表达的Treg细胞中诱导稳定表达荧光tdTomato)与XCR1-Venus/WT cDC1报告小鼠进行杂交。将OT-1 T细胞转移到这些小鼠中,在VV-OVA感染后第3天,OT-1 T细胞在富含DC的枢纽中停滞(图1O)。相比之下,在相同微环境中检测到的Treg细胞并未停滞,而是保持了平均速度为8 mm/min的随机迁移活动,就像在稳态期间一样。这种Treg细胞的行为与抗原特异性CD8 T细胞(迁移停滞)和抗原特异性CD4 T细胞(交替停止和移动)不同(图1P)。值得注意的是,在T细胞启动的初始阶段,即感染的第一天,Treg细胞也未迁移停滞(图S1,N和O)。作者考虑了只有Treg细胞的一个亚群可能被特异性吸引到CD8 T细胞被保留的亚滤泡枢纽的可能性。
3 CXCR3促进IL-2的获取并促进CD8 T细胞效应子的构建,发生在独立的启动阶段
在抗原特异性细胞相互作用期间,免疫突触的形成需要钙离子(Ca²+)的动员。相应地,T细胞从DCs的分离和细胞周期的启动依赖于T细胞受体(TCR)的脱敏,并且与受损的Ca²+信号传导相关。作者分离体内通过VV-GP激活的抗原特异性P14 T细胞,并在体外测量其对硫代硫酸钠的Ca²+流入反应(硫代硫酸钠会引发内质网储存耗竭,进而激活细胞外Ca²+流入)。在感染后24小时、48小时以及72小时分离的激活P14 T细胞显示出对硫代硫酸钠的Ca²+流入反应,这种反在添加细胞外Ca²+后进一步增加(图2A)。这些结果表明,至少有一部分T细胞在初始激活后保留了动员Ca²+的能力——暗示其对TCR刺激能够持续或再次获得反应性的潜力。这是作者观察到的第二次启动阶段中细胞停滞的关键,并且与其他研究中使用肽脉冲DCs的结果形成对比。为了进行体内分析,作者构建了表达遗传编码的Ca²+传感器GCaMP6F的P14 T细胞,并在第3天使用活体显微镜(IVM)进行分析。作者发现,在亚滤泡生态位中停滞的P14 T细胞显示出振荡的荧光信号,表明Ca²+通量(图2,B和C),这与TCR信号传导一致。
接下来,作者研究了哪种趋化因子受体可能参与将T细胞带到与DCs接近的位置,并促进更长期的接触。其中CXCR3在T细胞激活后的第2天至第3天表达上调。作者将WT[绿色荧光蛋白(GFP)]和CXCR3基因敲除(KO)OT-1 T细胞(用tdTomato标记)转移到WT受体中,用VV-OVA感染它们,并在3天后分析其在组织切片中的定位(使用共聚焦显微镜)。基于它们的空间积累或与淋巴结被膜的距离,WT和CXCR3 KO OT-1 T细胞在引流淋巴结中的整体分布相似,但它们的动态行为可能不同。因此,作者在相同条件下应用IVM直接比较WT和CXCR3 KO OT-1 T细胞的迁移行为,发现CXCR3 KO和WT细胞聚集在一起;然而,迁移停滞的CXCR3 KO细胞的比例低于WT OT-1 T细胞(图2,D到H)。同样,WT OT-1 T细胞的平均速度和轨迹位移长度也低于CXCR3 KO OT-1 T细胞(图2,E和F)。此外,少数停滞的CXCR3 KO细胞平均停滞时间更短(图2G)。作者发现,在检测到OT-1 T细胞簇的区域,CXCR3 KO T细胞的数量比其WT对应细胞少约三倍(图2H)。IVM分析显示,WT与CXCR3 KO OT1 T细胞的总体丰度相似(图2H)。这些结果说明CXCR3并不调节激活的CD8 T细胞的整体分布,而是影响它们在特定亚滤泡枢纽中的募集和/或保留。
为此作者重新分析了WT和CXCR3 KO OT-1细胞在组织切片上的分布。作者将WT和CXCR3 KO OT-1 T细胞转移,并用VV-OVA和VV-GP感染小鼠。在分析前8小时,作者转入了一组新的抗原特异性幼稚SMARTA T细胞作为细胞探针,以揭示激活的OT-1 T细胞被保留的生态位(图2I)。具体来说,作者使用激活的(CD69+)SMARTA T细胞作为参考点,并在淋巴结组织切片的共聚焦图像上测量WT和CXCR3 KO OT-1 T细胞的距离。与IVM结果相符,作者发现CXCR3 KO T细胞比其WT对应细胞参考点更远(图2J)。这些结果表明,在第二次启动阶段,CXCR3促进了激活的CD8 T细胞在特定亚滤泡枢纽中的获取和细胞停滞。
接下来,为了探究感染后第3天CXCR3依赖的相互作用期间传递了哪些信号,作者在感染第8天分析CD8 T细胞,发现此时CD8 T细胞已完全分化为效应T细胞(Teff)。与已发表的结果一致,作者发现CXCR3 KO CD8 T细胞未能在感染后第8天在淋巴结中通过Killer细胞凝集素样受体G1(KLRG1)表达分化为Teff(图2,K到M)。IL-12作为信号3细胞因子,指导CD8 T细胞分化,并且能够产生IL-12的细胞可以通过使用将黄色荧光蛋白(YFP)表达与p40亚基(IL-12p40-IRES-eYFP)偶联的小鼠来检测。在IL-12p40-IRES-eYFP小鼠中,表达YFP的cDC1s在相关生态位中富集,并在第3天与激活的OT-1相互作用(图S2,H到J)。作者前期工作已经证明cDC1s对于CD8 T细胞记忆构建至关重要,但并非Teff细胞分化所必需。对XCR1WT/DTR(DTR,白喉毒素受体)小鼠进行白喉毒素处理效果理想,导致cDC1s减少了98%。然而,cDC1耗竭并未阻止CD8 T细胞在第3天停滞,表明在这一启动阶段,cDC1和IL-12信号传导是冗余的。
因此,作者将注意力转向调节CD8 T细胞分化的重要因子IL-2,并探究这种细胞因子是否在通过CXCR3获取的细胞枢纽中被感知。作者将WT和CXCR3 KO OT-1 T细胞共同转移到WT受体中,用VV-OVA感染它们,并在感染后第3天,通过流式细胞术分析信号转导子和转录激活因子5(pSTAT5)的磷酸化,作为IL-2信号传导的衡量标准。作者发现与WT OT-1 T细胞相比,CXCR3 KO细胞中的pSTAT5水平降低(图2,N和O)。
CD8 T细胞的增殖能力与T细胞因子1(TCF1)蛋白的表达相关,该蛋白由Tcf7基因编码。因此,作者探究了是否主要表达低或高TCF1水平的CD8 T细胞母细胞获得了对富含IL-2的生态位的访问。在感染后第3天,作者发现,在转移的P14 TCF7GFP细胞中,pSTAT5阳性细胞的比例在TCF1低细胞中高于TCF1高细胞,这与TCF1低细胞中CXCR3、CD25和TCRβ的高表达水平一致(图2,P到R)。然而,由于作者无法在IVM期间区分转移的P14 TCF7GFP细胞中的TCF1低和TCF1高细胞,因此尚需确定在这一独特的启动阶段,CD8 T细胞在哪个特定的分化阶段重新与DCs相互作用。总的来说,作者的结果表明,CD8 T细胞表达的CXCR3通过在第二次启动阶段优化对亚滤泡生态位中IL-2的访问,促进了Teff的构建。
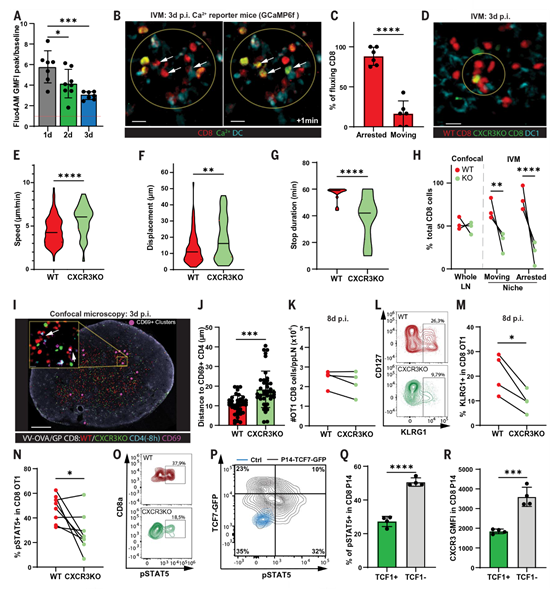
图2:CXCR3提供对IL-2的访问,并促进CD8 T细胞效应子的构建,发生在独立的启动阶段
4 Treg细胞动态调控CD8 T细胞效应子的构建,但独立于CXCR3
Treg细胞通过限制IL-2来部分调控抗病毒适应性免疫反应,从而抑制CD8 T细胞的扩增和Teff分化。利用Foxp3-DTR小鼠耗竭Treg细胞后,在VV-OVA感染后第8天,病毒特异性、VVB8R四聚体结合的CD8 T细胞中表达Teff细胞标记物KLRG1的比例增加(图3A)。同时,如果在初始启动阶段后24小时耗竭Treg细胞,这些病毒特异性CD8 T细胞的绝对数量也会增加(图3B)。当在感染后48小时耗竭Treg细胞时,也观察到了这种效应,表明Treg细胞的调控与CD8 T细胞在淋巴结中的第二次启动阶段在时间上有所重叠。一致地,感染后48小时耗竭Treg细胞会导致在VV-OVA感染后第3天过继转移的OT-1 T细胞中pSTAT5水平增加(图3C)。这些数据说明,Treg细胞通过减少IL-2的可用性来抑制CD8 T细胞的增殖和分化。值得注意的是,与OT-1 T细胞相比,Treg细胞的pSTAT5水平较低,这与其在富含IL-2的生态位中停留时间较短的结果一致(图1,O和P)。
由于抗病毒CD8 T细胞需要CXCR3才能与辅助性CD4 T细胞在亚滤泡簇中共定位,作者研究了Treg细胞表达CXCR3是否是限制CD8 T细胞分化的必要条件,通过感染CXCR3ΔFoxp3(Foxp3Cre × Cxcr3fl/fl)小鼠进行实验。与WT小鼠相比,感染后第8天,表达KLRG1的CD8 T细胞的比例以及VVB8R四聚体结合的CD8 T细胞的总数均未发生改变(图3,D和E)。因此,这些数据表明,Treg细胞对病毒特异性CD8 T细胞反应的调控并不需要CXCR3的表达。只有部分Treg细胞磷酸化STAT5的观察结果,提示在感染后第3天,特定亚群的Treg细胞可能在这些相互作用中调控CD8 T细胞。
为了解析Treg细胞的异质性和激活程序,作者从未感染和VV-OVA感染的Foxp3-GFP小鼠的淋巴结中分选Treg细胞,然后进行标记和混合,以便进行scRNA-seq。在稳态下,作者检测到三个不同的Treg细胞簇,相应地,在感染后也检测到三个簇,以及一个混合簇(图3F)。作者将其中一个簇命名为效应Treg(eTreg)细胞,其特征是表达Cd44、Icos、Tnfrsf9以及多种趋化因子受体,如Cxcr3和Ccr2(图3G)。在稳态下,该簇在与IL-2依赖通路相关的基因表达评分中最高(图3H)。另外两个独立的簇表达Sell(CD62L)、Ccr7和Klf2,并且IL-2信号评分较低,因此作者将其称为静止Treg(rTreg)细胞(图3,G和H)。最后,剩余的簇具有混合特征,其特征是表达Cd69和Nr4a1,表明存在活跃的TCR信号传导(图3G)。总体而言,感染后,Treg细胞的Ifit3和Socs1表达增加,说明IFN-γ和IL-2依赖通路的激活增加(图3,H到K)。值得注意的是,与IFN信号相关的基因表达相比,感染后IL-2信号相关基因表达的增加主要出现在rTreg细胞中(图3,G和H)。从成像(图1O)和功能数据(图3,A到E)来看,Treg细胞在感染后被全局激活,并独立于CXCR3进入相关生态位的途径。从功能上讲,Treg细胞似乎限制了IL-2的总体丰度,从而限制其可用性。相应地,在缺乏Treg细胞的情况下,CXCR3基因敲除(KO)OT-1 T细胞的Teff细胞构建得以恢复,这表现为在VV-OVA感染后,相对数量以及在较小程度上绝对数量的KLRG1表达细胞增加(图3L)。然而,IL-2的可用性在多大程度上受到Treg细胞在这些亚滤泡启动枢纽内外的调控仍不清楚。因此,作者得出结论,促进CD8 T细胞效应子构建的第二次启动阶段受到Treg细胞的限制,而Treg细胞动态地穿越生态位且独立于CXCR3。
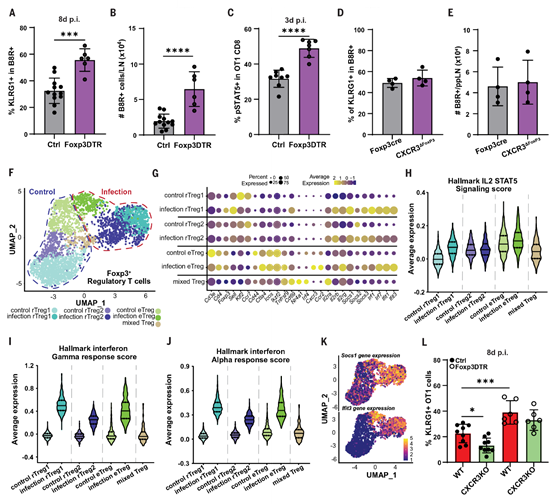
图3:Treg细胞动态调控CD8 T细胞效应子的构建,但独立于CXCR3
5 CD4 T细胞提供旁分泌IL-2以促进CD8 T细胞效应子的分化和扩增
为了确定第二次启动阶段中IL-2的细胞来源,作者构建了IL2ΔCD8(CD8aCre × Il2fl/fl)小鼠,并在VV-OVA感染后第8天分析了VVB8R四聚体结合的CD8 T细胞。在CD8 T细胞无法产生IL-2的小鼠中,作者没有观察到Teff构建的差异, CD8 T细胞数量和表达KLRG1及CX3CR1的细胞比例没有变化(图4,A到C)。
接下来,作者将注意力集中在CD4 T细胞上,认为它们可能是亚滤泡区域中传递的旁分泌IL-2的潜在来源。在通过抗体介导耗竭CD4 T细胞后,作者在感染后第3天观察到转移的OT-1 T细胞中pSTAT5水平降低(图4D)。然而,在其他研究中,CD4 T细胞耗竭对急性抗病毒CD8 T细胞反应的影响较小。这些观察结果与IL-2Rα缺陷(CD25 KO)OT-1 T细胞的CD8 T细胞扩增和效应分化显著减少形成对比(图4,E到G),这提示CD4辅助T细胞可能产生冗余的IL-2。
目前用于评估CD4 T细胞辅助作用的模型(例如MHC-II缺陷或抗体介导的CD4 T细胞耗竭)并不具有细胞类型特异性,会导致同时丢失具有抑制作用的CD4表达的Treg细胞。为了选择性地耗竭CD4辅助T细胞(CD4help),同时保持Treg细胞群完整,作者构建了CD4-DTR小鼠,其中DTR由两个loxP位点包围,并将其与Foxp3Cre ERT2小鼠杂交(此后称为CD4help-DTR)。经过他莫昔芬处理后,Cre介导的重组在Treg细胞中切除DTR,从而在白喉毒素(DT)给药后使它们免于细胞死亡(图4,H和I)。事实上,在VV感染后第8天,WT和CD4-DTR小鼠中VVB8R四聚体阳性的CD8 T细胞的绝对数量相似,这些小鼠中同时耗竭了辅助性和调节性CD4 T细胞。相比之下,使用CD4help-DTR小鼠选择性耗竭CD4辅助T细胞后,病毒特异性CD8 T细胞的数量减少了五倍以上(图4J)。此外,在CD4-DTR和CD4help-DTR小鼠中,KLRG1+或CX3CR1+ VVB8R四聚体阳性Teff细胞的频率降低(图4,K和L),并且它们的绝对数量在CD4help-DTR小鼠中进一步减少。这些数据确立了CD4辅助细胞已知记忆细胞功能外的在决定急性抗病毒CD8 T细胞反应数量中的作用。此外,这些结果还暗示Treg细胞可以在没有CD4辅助T细胞的情况下发挥抑制作用。
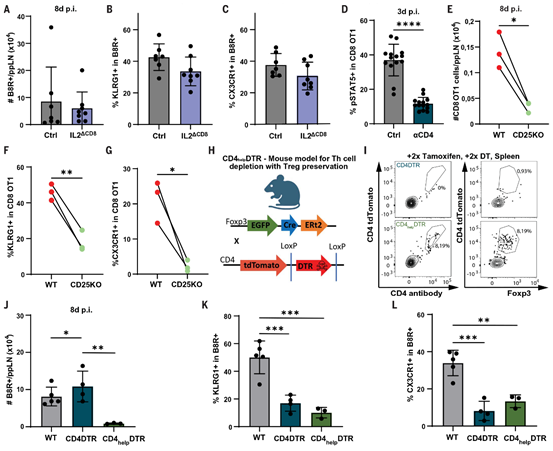
图4:CD4 T细胞提供旁分泌IL-2以促进CD8 T细胞效应子的分化和扩增
6 CD8 T细胞第二次启动阶段的亲和力选择
作者的研究结果揭示了一个独特的T细胞启动阶段,该阶段负责将CD4辅助T细胞产生的旁分泌IL-2传递给CD8 Teff,以调控其分化和扩增。这些发现与调节B细胞及其在生发中心反应期间的克隆选择以实现亲和力成熟的CD4 T细胞辅助功能存在相似之处。因此,作者研究了CD8 T细胞的第二次启动阶段是否也可能类似地用于选择高亲和力的CD8 T细胞克隆以进一步扩增。为了验证这一假设,作者将低亲和力的OVA257特异性CD8 T细胞(OT-3)与它们的高亲和力对应细胞(OT-1)的动态行为进行了比较。在VV-OVA感染后第1天,对淋巴结滤泡间区域(IFA)的活体显微镜(IVM)观察显示,在初始启动阶段,OT-1和OT-3 T细胞的迁移停滞并共同聚集(图5,A到D)。通过共聚焦显微镜对淋巴结切片的连续分析证实了这一结果,OT-1和OT-3 T细胞均表达CD69,说明这两种细胞通过TCR介导被激活(图5E)。
已有研究表明,T细胞受体(TCR)亲和力控制T细胞与树突状细胞(DC)相互作用的持续时间,作者也观察到T细胞与DC相互作用的动态存在细微差异(图5B)。然而,OT-1和OT-3细胞以相似的方式增殖(图5F),并在第3天在淋巴结中达到了相当的绝对数量(图5G)。
接下来,作者通过将OT-3 T细胞与OT-1 T细胞在同一个淋巴结中直接比较,分析了OT-3 T细胞在第二次启动阶段的动态行为。与OT-1 T细胞不同,低亲和力的OT-3 T细胞未能与DC进行长期重新相互作用(图5,I到L)。一致地,尽管CD25水平相似,但在感染后第3天,OT-3 T细胞中pSTAT5阳性的比例低于OT-1 T细胞(图5,M和N),证实了OT-3 T细胞对富含IL-2的生态位的访问受限。为了测试由这些生态位中的DC网络调控属于细胞内在还是细胞外在,作者在感染后第3天将幼稚的OT-1和OT-3 T细胞转移至小鼠体内,并分析它们的激活模式和定位。转移后8小时,作者通过CD69上调检测到幼稚OT-3 T细胞的强烈激活,尽管其频率低于OT-1 T细胞(图5O)。值得注意的是,在组织切片上,OT-1和OT-3 T细胞在淋巴结的相同生态位中共同聚集(图5P)。这些结果表明,低亲和力T细胞在第二次启动阶段未能积累相同水平的IL-2信号,这是由于它们在初始激活后基于TCR的迁移停滞能力发生了改变。与之相关的是,在感染后第8天检测到的OT-3 T细胞数量比OT-1 T细胞减少了10倍以上(图5Q)。同样,OT-3 T细胞的Teff构建,以表达KLRG1和CX3CR1的细胞比例为指标,也低于OT-1 T细胞(图5,R和S),并且其数量减少了100倍以上。Treg细胞耗竭并不能在感染后增加OT-3细胞的总数,但确实增加了其中Teff细胞的比例。综上所述,作者的数据与以下观点一致:第二次启动阶段的作用是选择高亲和力的CD8 T细胞克隆,以通过CD4 T细胞衍生的IL-2信号进一步扩增和分化。
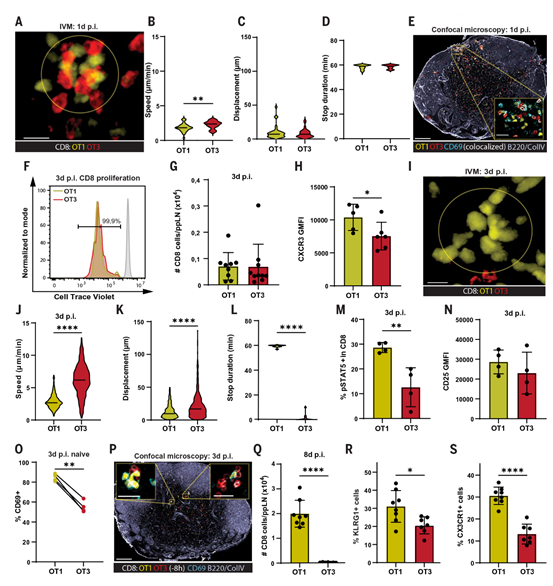
图5:在时间上分离的CD8 T细胞启动阶段的亲和力选择
结论:
综上所述,作者研究揭示了CD8 T细胞在免疫反应中的两个启动阶段,其中第二阶段通过CD4 T细胞提供的IL-2信号和Treg细胞的调节作用,选择性地促进高亲和力CD8 T细胞的扩增和分化,为疫苗和免疫疗法的开发提供了新机制和靶点。
实验方法:
活体成像技术(IVM),流式细胞术,细胞迁移模式分析,scRNA-seq,免疫荧光,细胞培养
参考文献:
Jobin K, Seetharama D, Rüttger L, Fenton C, Kharybina E, Wirsching A, Huang A, Knöpper K, Kaisho T, Busch DH, Vaeth M, Saliba AE, Graw F, Pulfer A, González SF, Zehn D, Liang Y, Ugur M, Gasteiger G, Kastenmüller W. A distinct priming phase regulates CD8 T cell immunity by orchestrating paracrine IL-2 signals. Science. 2025 Apr 11;388(6743):eadq1405. doi: 10.1126/science.adq1405. Epub 2025 Apr 11. PMID: 40208984.