






肝细胞BPGM诱导RET乳酸化和巨噬细胞重编程促进肝癌的发生
有氧糖酵解是癌症的标志,但糖酵解关键酶双磷酸甘油酸变位酶(BPGM)在肝细胞癌(HCC)进展中的作用尚不清楚。在本研究中,临床样本分析表明BPGM在HCC组织中表达上调,并与不良预后相关。肝细胞特异性Bpgm敲除显著减弱DEN诱导的小鼠HCC进展。空间转录组学和单细胞RNA测序显示,肝细胞特异性的Bpgm敲除减少了单核/巨噬细胞的浸润,并减少了肿瘤相关巨噬细胞的M2型极化。此外,BPGM过表达促进肝癌细胞的增殖和迁移,并促进细胞内乳酸的积累。液相色谱-串联质谱(LC - MS/MS)鉴定出RET原癌基因(RET)是介导BPGM对肝癌细胞作用的下游效应基因。BPGM促进P300介导的RET第549位赖氨酸(K549)的乳酸化,竞争性地抑制其泛素化,从而阻止RET蛋白的降解并增强其稳定性。肝癌细胞中的BPGM还通过乳酸分泌诱导巨噬细胞的组蛋白乳酸化和M2型极化。本研究揭示肝细胞中的BPGM可以通过增加恶性细胞中RET的乳酸化而增强其表达,并促进巨噬细胞的M2型极化,这两者都促进了HCC的进展。这些发现证实了BPGM可作为HCC的潜在治疗靶点。该研究于2026年3月发表在《Advanced Science》,IF:14.1。
技术路线

1.BPGM在肿瘤组织中的表达上调与HCC患者预后不良相关
我们首先查询了癌症基因组图谱(The Cancer Genome Atlas,TCGA)和基因表达汇编(Gene Expression Omnibus,GEO)数据库,以评估HCC进展期间的BPGM转录水平,发现与正常肝组织相比,BPGM mrna表达在HCC患者的肿瘤组织中显著升高(p < 0.001)(图1A,B)。我们队列的结果同样表明,在40例HCC患者的肿瘤组织中,BPGM的mRNA水平显著高于癌旁组织(p < 0.05)(图1C)。基于TCGA数据集,DeLong对相关受试者工作特征(ROC)曲线的检验显示,在区分HCC组织和正常肝组织方面,BPGM的曲线下面积(AUC)(0.8517)显著高于AFP (0.6160) (p < 0.001),证实了BPGM在诊断效能上显著优于AFP(图1D)。此外,我们对HCC组织和癌旁组织(n = 15)进行了免疫组化染色,以检测HCC患者BPGM的蛋白表达,发现BPGM在HCC组织中表达较高(p < 0.0001)(图1E)。对TCGA数据集的分析显示,高肿瘤分级(G3/G4)和晚期(III/IV)的患者BPGM mRNA水平升高(p < 0.05)(图1F,G)。然后,我们基于TCGA数据库将BPGM的表达与HCC患者的生存率进行了关联,并发现BPGM高表达的患者显示出更短的总生存期(OS)和无病生存期(DFS)(图1H,I)。这些数据证实了BPGM在肝癌组织中的转录和蛋白水平上过表达,与患者的不良预后相关,提示BPGM有潜力作为肝癌的诊断生物标志物。
为研究BPGM在不同细胞类型中的表达,我们分析了来自两个数据集的HCC样本的整合scRNA - seq数据,GSE149614(8个非肿瘤样本和10个肿瘤样本)和GSE202642(4个非肿瘤样本和7个肿瘤样本)。推断的拷贝数变异(CNV)分析验证了肿瘤细胞的存在。细胞比例如图1J所示。我们观察到BPGM在肿瘤组织的肝细胞中的表达显著高于非肿瘤肝组织(p < 0.0001)(图1K)。而在肿瘤组织中,恶性HCC细胞中的BPGM表达水平显著高于非恶性肝细胞(p < 0.0001)(图1L)。此外,与非肿瘤肝组织相比,肿瘤组织中内皮细胞、成纤维细胞和B/浆细胞中的BPGM表达水平也显著升高(图S1A-C),而在髓系细胞和T/NK细胞中未观察到这种差异(图S1D,E)。这些结果表明肝细胞特异性BPGM可能在HCC发病机制中作为肿瘤促进因子发挥作用。
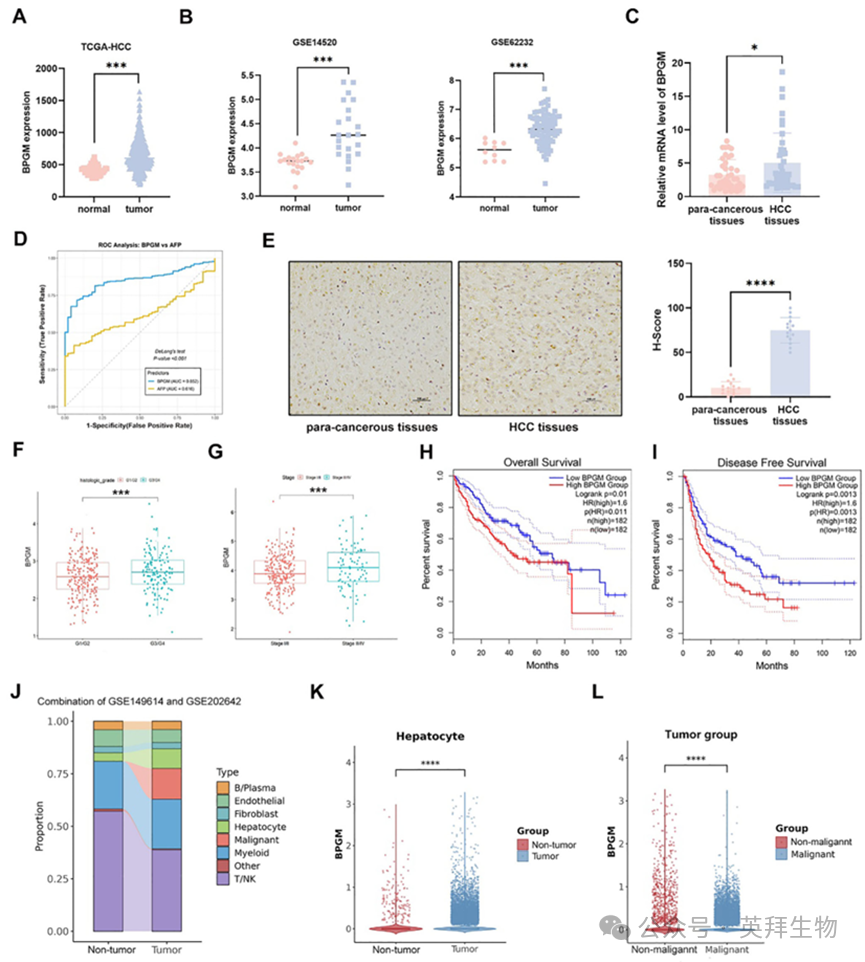
图1.BPGM在HCC组织中的表达水平上调
2.体内敲除BPGM抑制HCC肿瘤生长
为分析BPGM在肝癌发生中的作用,我们利用CRISPR‐Cas9和Loxp‐Cretechnologies构建了肝细胞特异性BPGM‐敲除小鼠模型(图S2A-C)。通过腹腔内注射烷化剂二乙基亚硝胺(DEN)诱导HCC小鼠模型(图2A)。与Bpgm‐CON小鼠相比,Bpgm‐CKO小鼠肝脏中Bpgm mRNA的表达显著降低(p < 0.01),而在心脏、大脑和肾脏中没有检测到差异(图2B,C)。这些结果证实了成功构建了肝细胞特异性Bpgm基因敲除小鼠模型。在Bpgm‐CON小鼠中可见大量不同大小的结节,占据了大部分肝脏表面积。在Bpgm‐CKO小鼠中,除了肝脏表面有一些结节外,肝脏在外观、颜色和硬度上几乎与正常肝脏相似(图2D)。总之,与Bpgm‐CON小鼠相比,Bpgm‐CKO小鼠的肝脏重量(p < 0.001)、最大肿瘤直径(p < 0.05)和肿瘤数量(p < 0.05)均显著降低(图2E-G)。肝脏苏木精-伊红(H&E)染色进一步证实了Bpgm‐CON小鼠中HCC的形态学特征(图2H)。此外,敲除Bpgm显著降低了血清和肝脏的总胆固醇(TC)水平(p < 0.001)(图2I,J)。与Bpgm‐CKO小鼠相比,Bpgm‐CON小鼠的α‐平滑肌肌动蛋白(α‐SMA)免疫组织化学染色(p< 0.0001)和天狼星红染色(p< 0.01)表明,Bpgm‐CON小鼠表现出更高程度的纤维化(图2K,L)。Ki‐67的免疫组织化学染色显示Bpgm‐CON小鼠的增殖能力显著高于Bpgm‐CKO小鼠(p < 0.001)(图2M)。综上所述,肝细胞特异性BPGM缺陷减缓了肝癌的进展,表明BPGM可能参与了肝癌的发生。
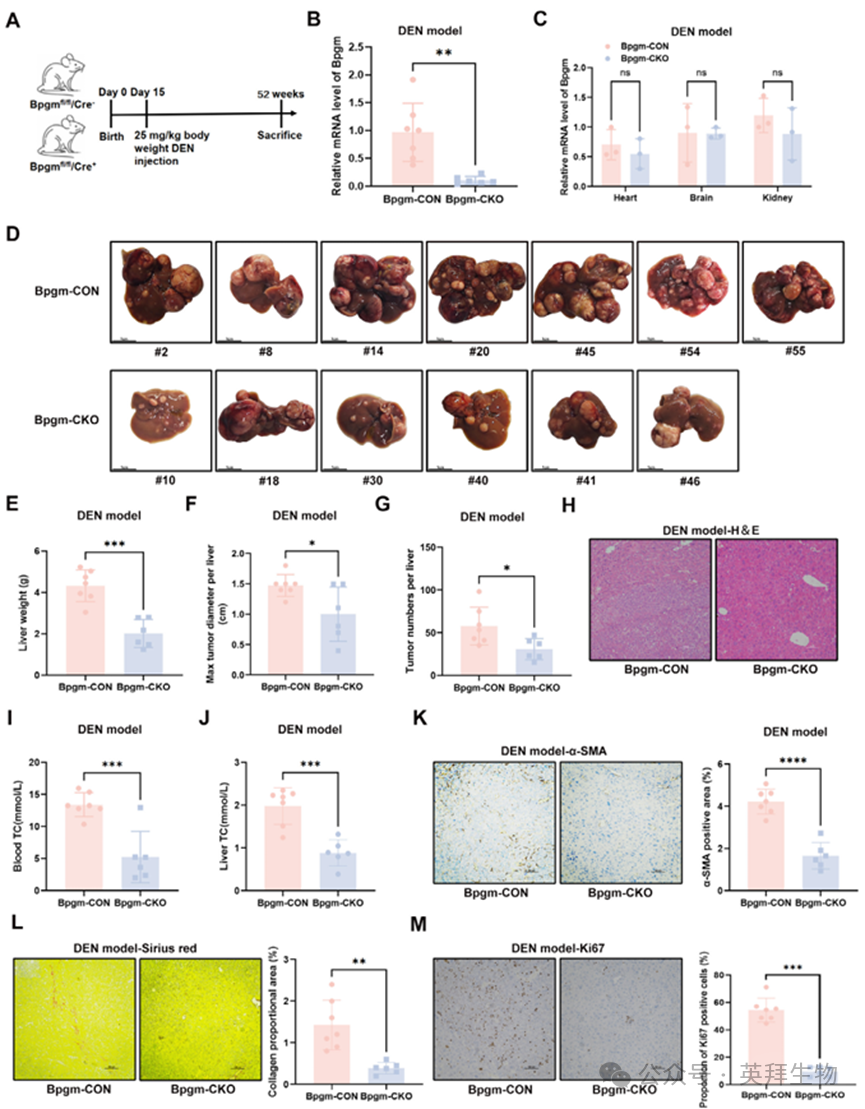
图2.体内敲除BPGM抑制HCC肿瘤生长
3.BPGM的表达显著增强单核/巨噬细胞在HCC TME中的浸润
为全面分析HCC肿瘤中不同细胞类型的空间分布,我们从Bpgm‐CON和Bpgm‐CKO小鼠的HCC中收集了6个肿瘤组织标本(每组n = 3),并利用Seurat将scRNA‐seq与空间转录组数据整合,从而重建空间单细胞图(图3A)。我们进一步发现,单核细胞/巨噬细胞是Bpgm‐CON小鼠肝细胞40µm半径内最丰富的细胞类型(p < 0.05)(图3B)。空间生态位分析显示,Bpgm‐CON小鼠中肝细胞富集生态位4中的单核细胞/巨噬细胞比例显著高于Bpgm‐CKO小鼠(p < 0.05)(图3C)。对肝细胞密度进行分层后,我们发现在肝细胞密度最高的30-70%区域,Bpgm‐CON小鼠的单核细胞/巨噬细胞比例显著高于Bpgm‐CKO小鼠(p < 0.05)(图3D;图S3)。综上所述,我们发现BPGM的表达促进了HCC TME中单核/巨噬细胞的浸润。
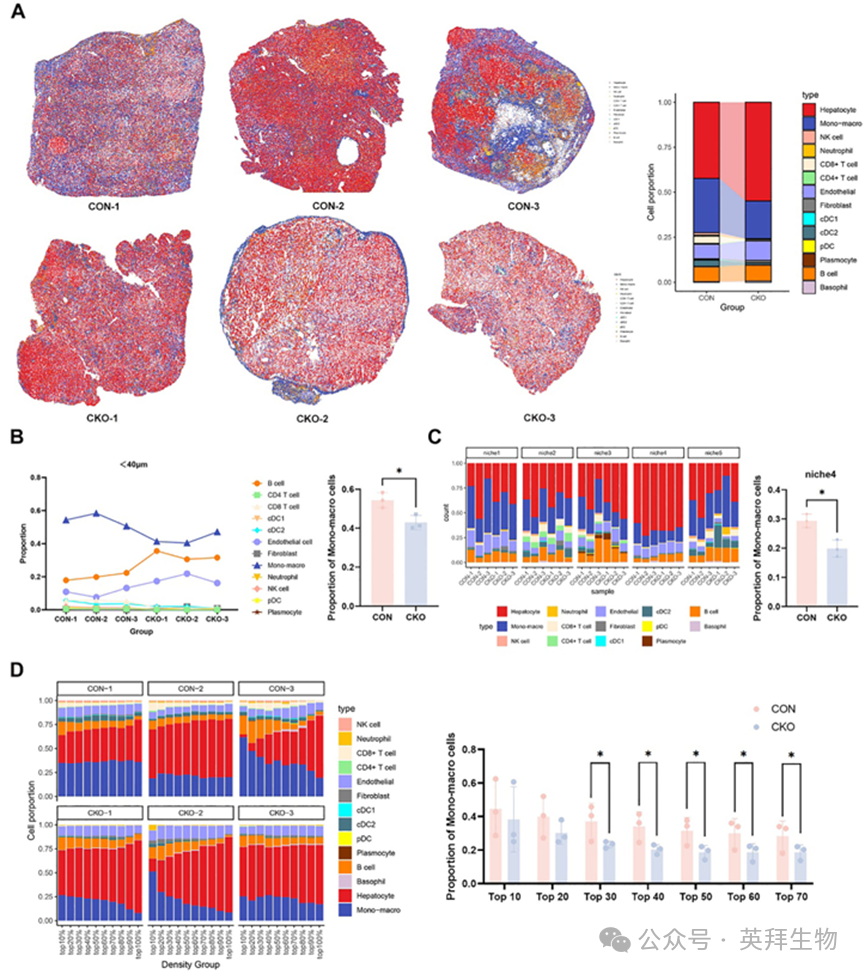
图3.单核/巨噬细胞在HCC TME中的空间分布
4.BPGM在单细胞水平诱导TAMs的M2极化
为在单细胞水平上评估HCC微环境中免疫细胞的可塑性和表型,我们对Bpgm‐CON和Bpgm‐CKO小鼠的肿瘤组织进行了scRNA‐seq。我们根据差异表达基因(DEGs)的富集分析将细胞群分为17个簇(图4A)。Bpgm‐CON和Bpgm‐CKO小鼠的统一流形近似和投影(UMAP)轮廓如图4B和图4C所示。比例分析显示Bpgm‐CKO小鼠中的恶性细胞(来源于肝细胞)和巨噬细胞的百分比降低(图4D)。此外,增殖途径的效应因子,包括Myc, Met, Jun, Ccnd1, Fos和Axin2,在Bpgm‐CON小鼠的恶性肝细胞中高表达(p < 0.001)(图4E)。随后,我们对巨噬细胞进行了亚聚类分析,确定了11个不同的亚群(图4F)。使用M2标记Mrc1,我们定义了两个主要人群:M2极化的肿瘤相关巨噬细胞(M2 TAMs)和非M2 TAMs(图4G-H)。值得注意的是,与Bpgm‐CKO小鼠相比,Bpgm‐CON小鼠表现出M2 TAM群体的显著扩增和M2 TAM中Mrc1的表达增强(p < 0.05)(图4I-J)。同样,肝组织免疫荧光结果显示,Bpgm‐CON小鼠中CD68+/CD206+ M2巨噬细胞数量增加(图4K)。综上所述,这些发现表明肝癌细胞中的BPGM驱动恶性细胞(来源于肝细胞)和M2 TAMs的协同激活。
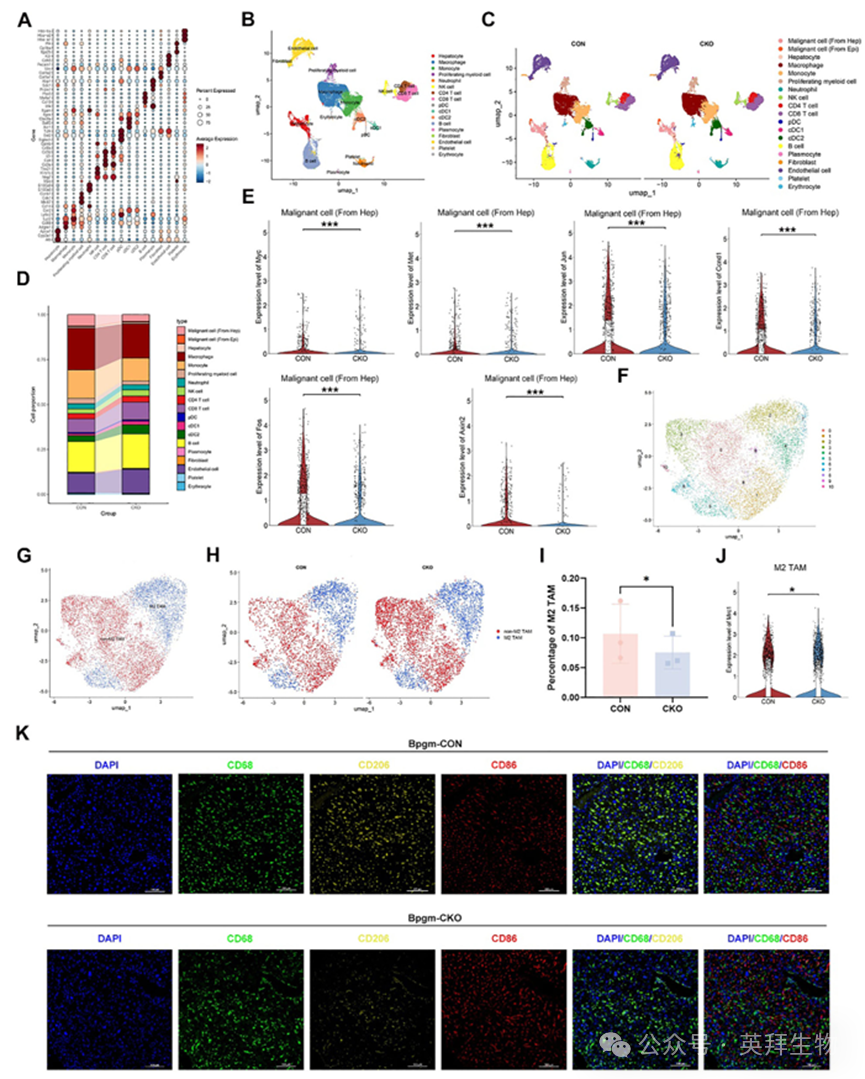
图4.肝癌小鼠Bpgm‐CON(n = 3)和Bpgm‐CKO(n = 3)的单细胞测序分析
5.BPGM在体外促进肝癌细胞增殖和迁移
接下来,我们构建了过表达BPGM的肝癌细胞系Huh7和PLC/PRF/5来研究BPGM的功能。我们在RNA和蛋白水平验证了BPGM的成功过表达(图5A,B)。CCK‐8实验和克隆形成实验的结果表明,过表达BPGM显著提高Huh7和PLC/PRF/5细胞的增殖速率(图5C-F)。伤口愈合和Transwell实验表明,BPGM的过表达显著增强了肝癌细胞的迁移能力(p < 0.05)(图5G-J)。此外,我们使用BPGM特异性siRNA来敲低HCC细胞系中的基因表达(图S4A,B)。我们发现通过si - BPGM - 3敲低BPGM显著降低了HCC细胞的增殖、迁移和集落形成能力(图S4C-J)。另一个针对不同位点的siRNA (si‐BPGM‐2)敲低BPGM也显示出相同的表型变化(图S5A-C)。综上所述,这些发现表明,靶向BPGM显著影响肝癌细胞的恶性表型,包括增殖和迁移。
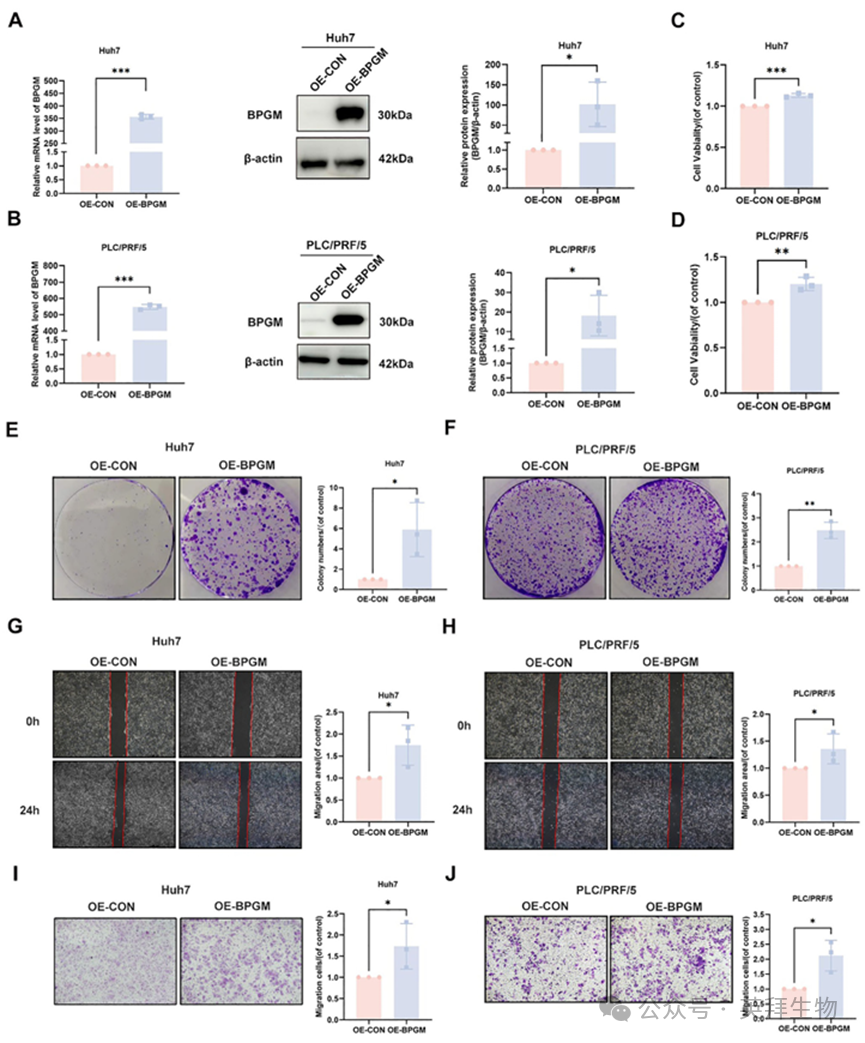
图5.BPGM过表达促进HCC细胞增殖、迁移和集落形成能力
6.BPGM与HCC细胞乳酸生成和蛋白乳酸化呈正相关
Bpgm‐CON和Bpgm‐CKO小鼠恶性细胞(来自肝细胞)转录组的基因集富集分析(GSEA)显示,乳酸代谢(p = 0.049)和乳酸化(p = 0.034)的生物过程显著富集(图6A,E)。此外,乳酸相关基因(Ldha, Pkm和Hif1α)在Bpgm‐CON小鼠的恶性细胞(来自肝细胞)中上调(p < 0.001)(图6B-D)。在乳酸化相关基因(Ep300, Aars1和Hk2)中也观察到类似的发现(p < 0.001或p < 0.05)(图6F-H)。来自GSE149614数据集的scRNA‐seq数据还表明,HCC肿瘤组织中的肝细胞表现出较高的LDHA、PKM和SLC2A1水平(p < 0.0001)(图S6A-C)。一致地,乳酸相关的信号通路在TCGA数据库中BPGM高表达的HCC患者组中显著富集(图6I)。鉴于糖酵解在癌症中的显著上调,并且乳酸是糖酵解的关键代谢物,探索乳酸相关信号在HCC中的参与是很重要的。体外实验表明,在BPGM过表达的PLC/PRF/5细胞中观察到细胞内乳酸浓度显著增加(p < 0.001)(图6J)。此外,相关分析显示,在TCGA数据库中371例HCC患者的肿瘤组织中,BPGM和EP300的表达呈显著正相关(r = 0.47, p < 0.0001)(图6K)。这些结果表明,BPGM mRNA水平与乳酸和泌乳密切相关。与Bpgm‐CON小鼠相比,Bpgm‐CKO小鼠肝脏中的泛乳酸化水平显著降低(图6L),而在Bpgm‐过表达的PLC/PRF/5细胞中,细胞内的蛋白质乳酸化水平也增加(图6M)。此外,我们在过表达BPGM的PLC/PRF/5细胞中进行了IP实验,以富集BPGM介导的乳酸化下游靶蛋白。在免疫沉淀的SDS - PAGE后,将凝胶切断并用硝酸银染色。免疫沉淀的Western blot实验表明,与OE‐CON‐Lac组相比,OE‐BPGM‐Lac组的乳酸化水平上调。具有乳酸化作用的差异蛋白的分子量集中在130-180 kDa范围内,表明BPGM可能靶向HCC细胞内的非组蛋白(图6N)。这些结果表明,BPGM促进肝癌细胞的乳酸生成和乳酸化。
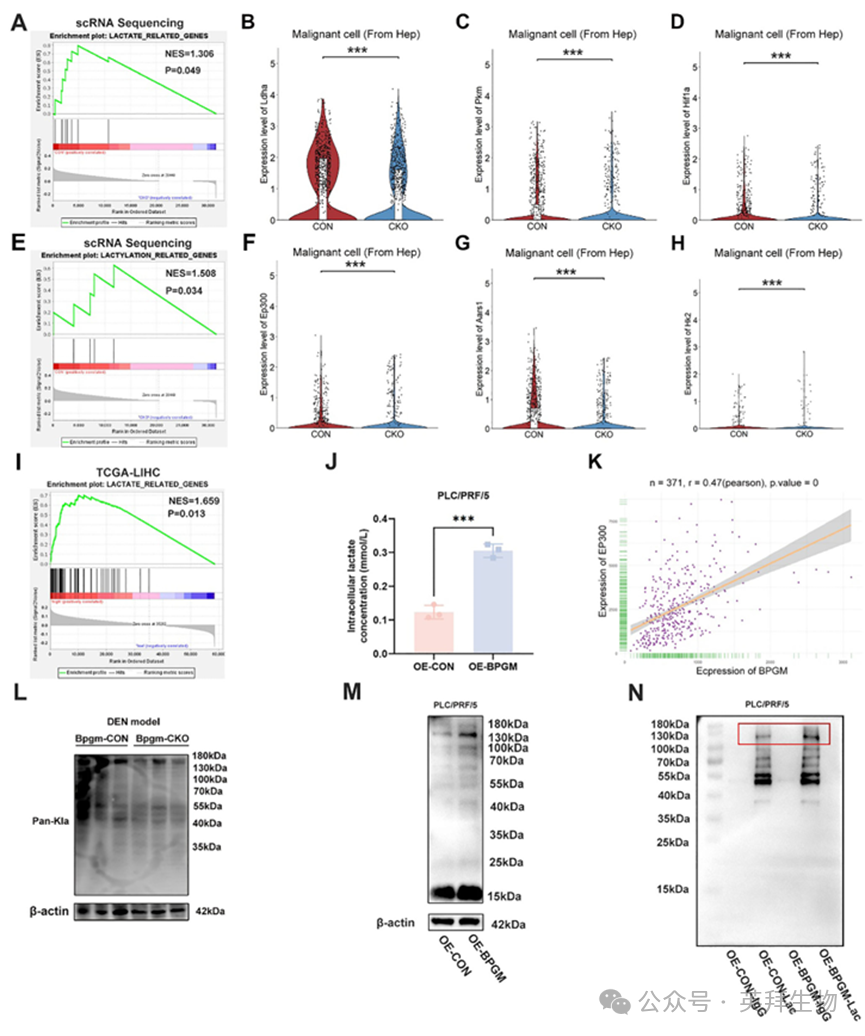
图6.BPGM与HCC细胞乳酸生成和蛋白乳酸化呈正相关
7.BPGM调节HCC细胞中RET - K549的乳酸化和泛素化
接下来,我们筛选并鉴定了BPGM介导的乳酸化的下游靶蛋白。将用乳酸化蛋白抗体免疫沉淀的蛋白质进行LC‐MS/MS分析。程序包括蛋白质提取、肽酶消化、质谱分析和数据分析(图7A)。图7B显示了一些在过表达BPGM的细胞中上调的顶级蛋白。在肝癌细胞中受BPGM影响的差异表达蛋白中,我们重点关注了ret原癌基因(RET),根据其在肝癌组织中的差异表达,RET可能是BPGM的一个重要下游蛋白(图7C)。我们的免疫共沉淀(co - IP)结果验证了BPGM促进RET的乳酸化(图7D)。在过表达BPGM的细胞中,RET的表达水平显著升高(图7E)。Bpgm‐CKO小鼠的肝组织显示RET蛋白表达降低(p < 0.05)(图7F),这与体外结果一致。由于泛素化影响蛋白质的降解,我们假设BPGM介导的乳酸化可能与泛素化竞争来阻止RET的降解。使用DeepKla和GPS‐Uber网站来预测RET上潜在的乳酸化和泛素化位点,我们确定了三个候选位点,并预测了这两种修饰(图7G)。为了确定RET上的潜在修饰位点,我们评估了三个位点(RET‐K523, RET‐K549和RET‐K965)的突变所获得的泛素化和乳酸化水平。首先,我们验证了RET‐WT、RET‐K523R、RET‐K549R和RET‐K965R的过表达效率(图7H)。Co‐IP结果显示RET‐K549R消除了RET的乳酸化和泛素化(p < 0.0001),而RET‐K523R和RET‐K965R突变对乳酸化和泛素化水平没有显著影响(图7I,J)。此外,用2‐脱氧‐D‐葡萄糖(2‐DG,一种葡萄糖抑制剂)处理PLC/PRF/5细胞以减少乳酸的产生,结果显示乳酸化减少,同时泛素化增加(图7K,L)。P300是一种潜在的乳酸化writer 蛋白,P300缺失会降低组蛋白乳酸化。我们使用siRNA敲低PLC/PRF/5细胞中P300的表达(图7M)。P300沉默后,蛋白质乳酸化水平降低(图7N), RET的泛素化显著上调(图7O)。综上所述,这些结果表明BPGM可以促进P300介导的RET K549位点的乳酸化和抑制RET泛素化,从而抑制其降解,最终导致RET蛋白水平的上调。
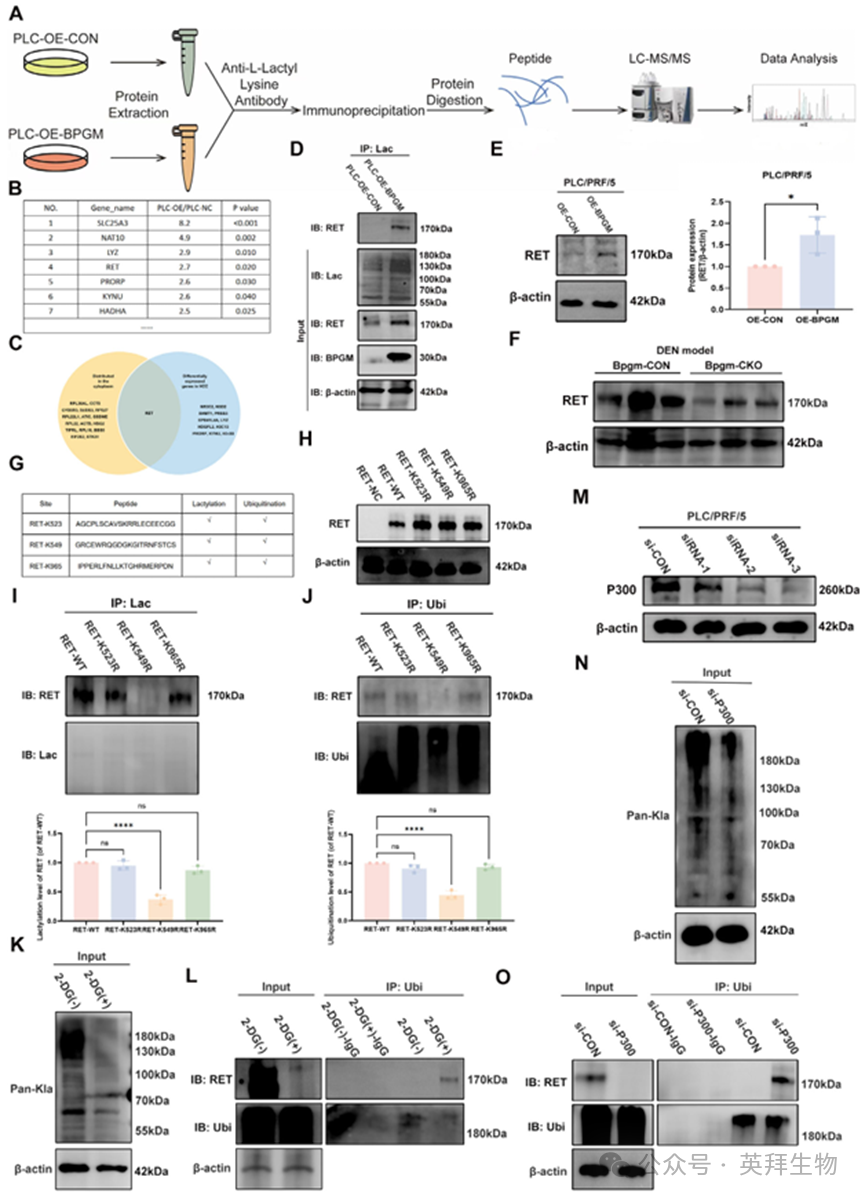
图7.BPGM调节HCC细胞中RET - K549的乳酸化和泛素化
8.RET介导BPGM诱导的HCC细胞增殖和迁移
考虑到BPGM可以促进RET蛋白的表达水平,我们研究RET是否可以介导BPGM在HCC进展中的功能。我们在过表达BPGM的PLC/PRF/5细胞中沉默RET基因。通过CCK - 8, wound - healing和Transwell实验,我们发现RET的敲低可以显著减弱BPGM诱导的肝癌细胞的增殖和迁移能力(图8A-C)。RET过表达可以恢复BPGM敲低导致的肝癌细胞增殖和迁移能力下降(图8D-F)。以上结果表明,BPGM通过促进RET的表达促进肝癌细胞的增殖和恶性表型。
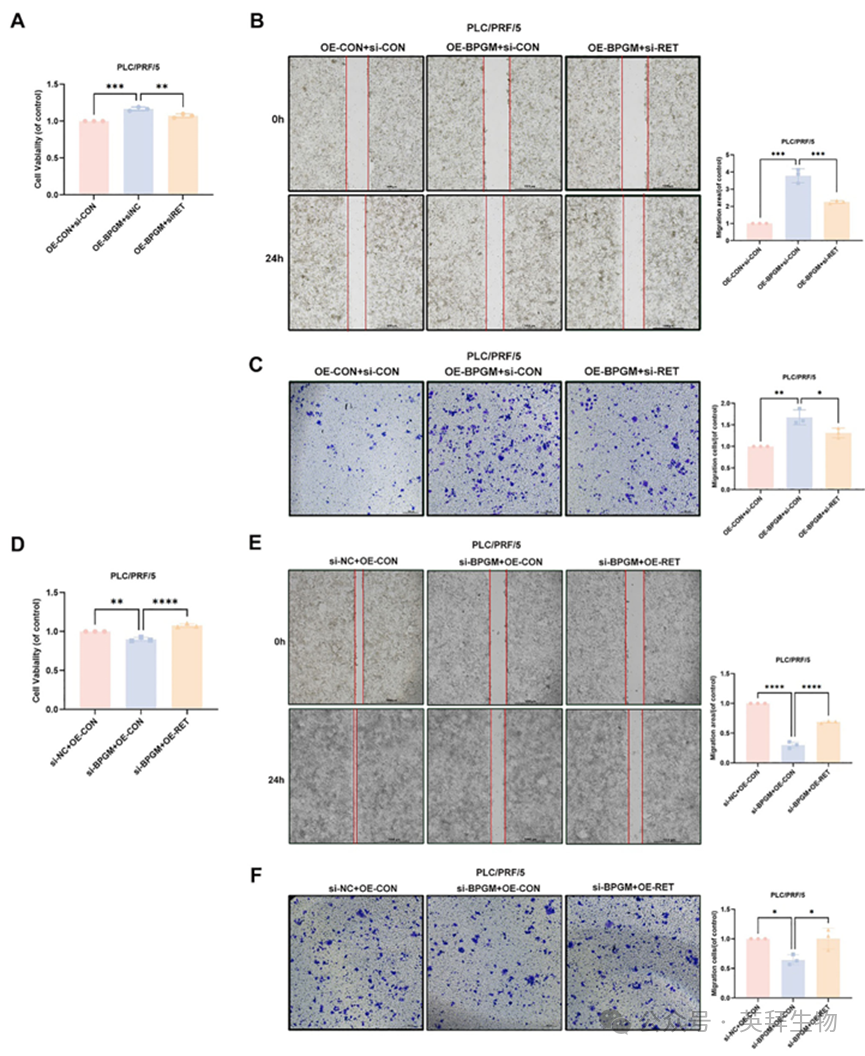
图8.RET在BPGM介导的HCC细胞增殖和迁移能力中的作用
9.BPGM在HCC细胞中促进巨噬细胞M2极化
我们进一步在体外验证了BPGM介导的HCC进展表型和巨噬细胞之间的相关性。与PLC‐OE‐CON组相比,PLC‐OE‐BPGM过表达(PLC‐OE‐BPGM)后PLC/PRF/5细胞上清液中的乳酸浓度显著升高(p < 0.001)(图9A)。基于这一现象,我们收集了过表达和未过表达BPGM的PLC/PRF/5细胞的上清液,然后用这些上清液处理由THP - 1细胞分化而来的M0巨噬细胞(图9B)。共培养后,western blot结果显示OE‐BPGM组巨噬细胞中约15 kDa的蛋白乳酸化水平显著上调(图9C),表明乳酸可能通过组蛋白乳酸化促进M2型巨噬细胞极化。利用TIMER和MCPCOUNTER算法分析TCGA队列中HCC组织中BPGM表达水平与微环境中巨噬细胞的相关性(图9D,E)。使用CIBERSORT‐ABS和QUANTISEQ算法的检测也表明,肝癌组织中BPGM的表达水平与M2巨噬细胞浸润呈正相关(图9F,G)。与OE‐CON组相比,在OE‐BPGM肝癌细胞上清液孵育的THP‐1细胞分化的M0巨噬细胞中,M2巨噬细胞标志物(MRC‐1、IL‐10、ARG‐1和CD163)的mRNA表达水平显著上调(p < 0.01或p < 0.05)(图9H-K)。CD206的蛋白表达水平显著上调(p < 0.01)(图9L)。流式细胞术结果进一步证实,OE‐BPGM组中CD68+CD206+ M2巨噬细胞的比例显著高于OE‐CON组(p < 0.05)(图9M)。此外,用MCT特异性抑制剂α‐氰基‐4‐羟基肉桂酸(CHC)和上述PLC‐OE‐BPGM细胞上清液孵育THP‐1细胞分化的M0巨噬细胞。共培养模型的示意图提供于图9N中。CHC降低了BPGM过表达的HCC细胞的上清液诱导的M0巨噬细胞中CD68+CD206+ M2巨噬细胞的比例(图9O-Q),减少了细胞内乳酸的积累,减少了M2标志物(CD163, IL - 10)的mRNA水平。所有结果表明,CHC介导的乳酸转运抑制显著地消除了BPGM过表达PLC/PRF/5细胞的上清液诱导的M2极化促进作用。
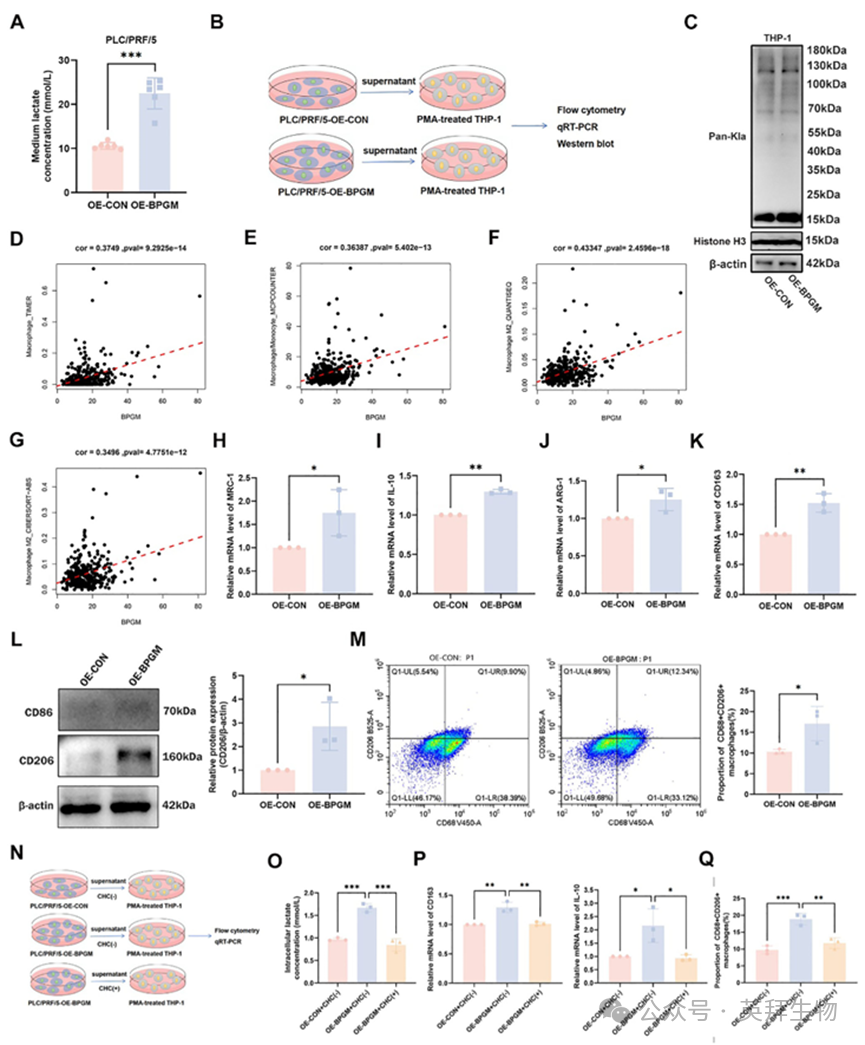
图9.BPGM过表达促进微环境M2巨噬细胞极化
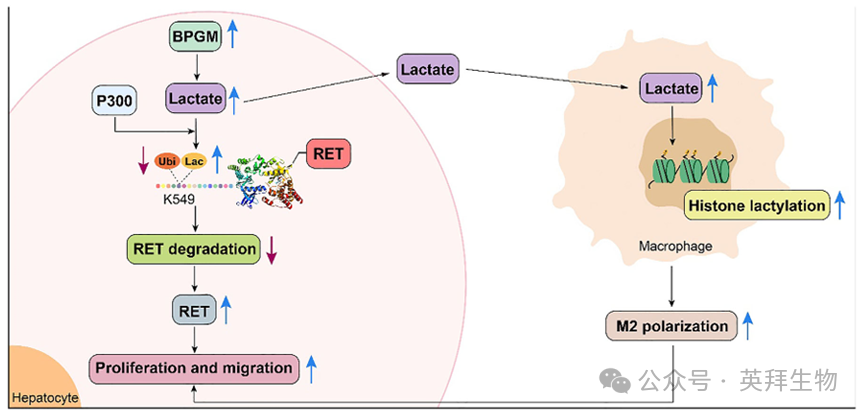
图9.机制图
结论
综上所述,本研究揭示了BPGM在促进肝癌发生中的新作用。BPGM诱导RET蛋白K549位点的乳酸化,竞争性抑制该位点的泛素化。这增加了RET的表达水平,介导了BPGM促进肝癌细胞增殖和迁移。此外,肝癌细胞中的BPGM促进了TME中乳酸的积累,增加了巨噬细胞中组蛋白的乳酸化水平,诱导了巨噬细胞的M2极化,这也可能促进了肝癌的发生(图10)。这些数据为肝癌发生的分子机制提供了新的见解,并提示BPGM可能是一个有希望的肝癌治疗靶点。
参考文献
Zhang J, Shi L, Lin L, Zhang Y, Zhang M, Wu L, Xia Y, Zhang Y, Han P, Zhuang L, Shi L. Hepatocyte BPGM Induces RET Lactylation and Macrophage Reprogramming to Promote Tumorigenesis in Hepatocellular Carcinoma. Adv Sci (Weinh). 2026 Mar;13(16):e18180. doi: 10.1002/advs.202518180. Epub 2026 Jan 9. PMID: 41514495; PMCID: PMC13042787.