






紫杉化疗通过STING介导的内质网应激和HMGB1分泌促进对TIM-3检查点阻断的反应
免疫检查点抑制剂正越来越多地与化疗方案结合使用,但这些组合成功或失败的原因尚不明确。在以往的研究中,我们描述了阻断 TIM-3 如何通过 HMGB1 依赖 DNA 摄取促进树突状细胞的激活,从而在与紫杉醇联合使用时具有疗效。本研究显示,肿瘤细胞释放 HMGB1 对于与 TIM-3 阻断结合紫杉醇、多西他赛、氟尿嘧啶及照射的组合疗效是必要的。紫杉烷治疗期间的 HMGB1 释放是一种涉及核外流的主动过程,涉及 Toll 样受体 4(TLR4)依赖的活性氧物种生成、DNA 损伤以及多(ADP-核糖)聚合酶活化。DNA 损伤促进细胞质双链 DNA(dsDNA)的积累,激活 cGAS-STING 通路;然而,紫杉类药物无法诱导 I型干扰素。相反,STING 激活促进内质网(ER)应激和溶酶体胞吐作用,推动 HMGB1 的分泌。因此,非典型的 STING 信号对紫杉类药物的反应可以促进化疗免疫疗法的疗效。这篇文章于2026年5月6日发表于《Cell Reports Medicine》期刊上,IF:10.6。
研究技术路线:
主要实验结果:
紫杉烷类药物促进HMGB1释放并增强TIM-3阻断疗效
我们首先探究哪些细胞毒性治疗可在体外诱导PyMT‑B6 肿瘤细胞释放 HMGB1。商品化 ELISA 试剂盒结果波动大,无法稳定检测细胞培养上清中的 HMGB1,因此改用免疫荧光共聚焦显微镜,在各药物 24 小时半数致死剂量(LD₅₀)下检测细胞内 HMGB1 含量。
经紫杉醇(PTX)及其半合成衍生物多西他赛(DTX)处理后,细胞核内 HMGB1 几乎完全消失(图 1A)。5‑氟尿嘧啶(5‑FU)的活性代谢产物 FdUMP或 24 Gy 放疗也可导致 HMGB1 显著减少;而阿霉素、吉西他滨、顺铂、环磷酰胺则无此效应。为排除单克隆抗体无法识别翻译后修饰 HMGB1 的干扰,我们构建了稳定表达 HMGB1‑ZsGreen 融合蛋白的 PyMT‑B6 细胞,验证了上述结果,且 HMGB1‑ZsGreen 的丢失在 8 小时达到峰值(图 1B)。值得注意的是,人三阴性乳腺癌(TNBC)细胞 MDA‑MB‑231 在 PTX、DTX 或 FdUMP 处理后也出现类似的 HMGB1 丢失。HMGB1 减少并非由基因转录下调所致,蛋白免疫印迹显示 PTX 或 DTX 处理后细胞上清中 HMGB1 显著增加。
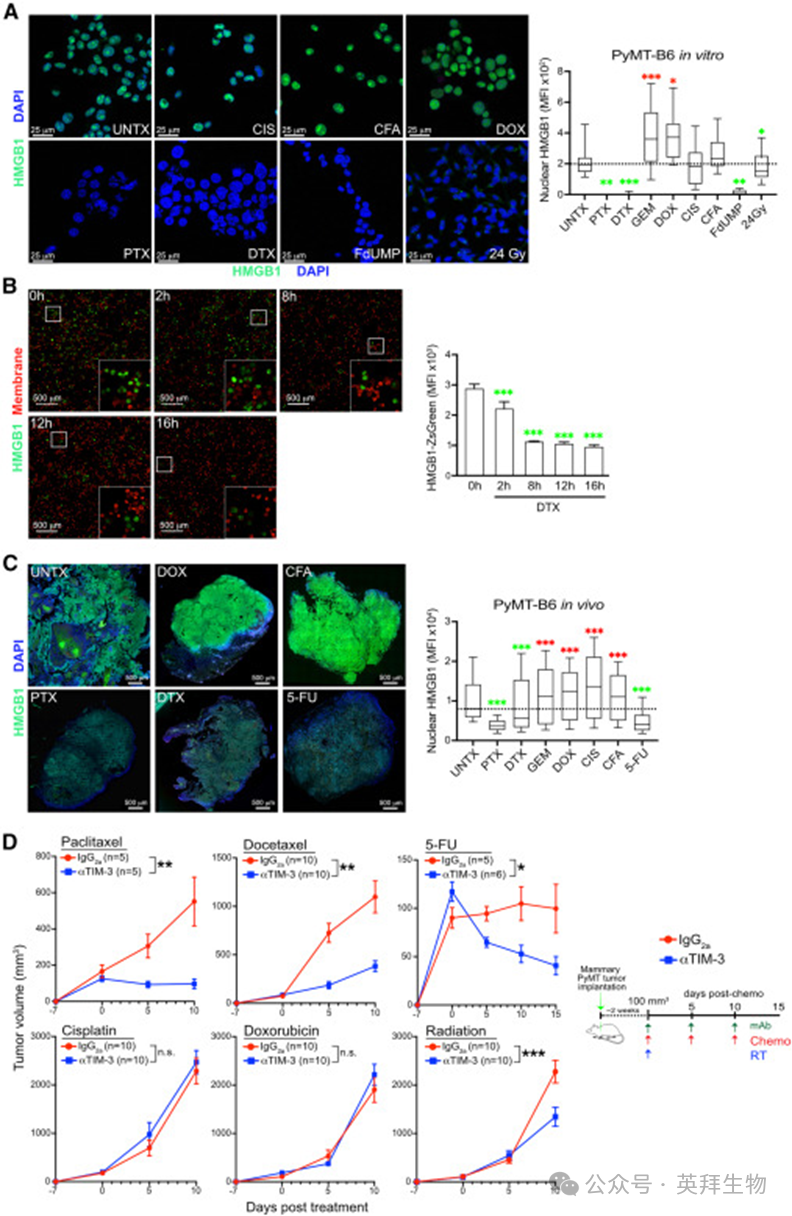
图1. 紫杉烷类药物促进HMGB1释放并增强对TIM-3 阻断的响应
随后我们在原位 PyMT‑B6肿瘤模型中验证体外结果,通过分析全肿瘤切片中单个细胞核内 HMGB1 的荧光强度判断其释放情况。PTX、DTX 与 5‑FU 处理组小鼠的肿瘤细胞核内 HMGB1 显著降低(图 1C);而 DNA 损伤类化疗药物吉西他滨、环磷酰胺、阿霉素和顺铂则使核内 HMGB1 增加。顺铂组的结果与 HMGB1 对铂‑DNA 加合物的高亲和力一致,但与经典免疫原性细胞死亡(ICD)及 DNA 烷化剂诱导 HMGB1 被动核外移的报道相悖。关键在于,PTX、DTX、5‑FU 与放疗诱导的 HMGB1 释放,与抗 TIM‑3 抗体联合这些细胞毒性治疗的增效效果完全对应;而抗 TIM‑3 无法增强顺铂或阿霉素的疗效(图 1D)。综上,肿瘤细胞的 HMGB1 释放与乳腺癌临床前模型中 TIM‑3 阻断联合化疗的疗效正相关。
紫杉烷治疗中 HMGB1 的主动分泌依赖 TLR4
细胞毒性治疗导致的HMGB1 丢失通常被认为是细胞膜破裂后的被动释放,但本研究中肿瘤活细胞区仍出现明显HMGB1 丢失(图 1C),且封闭膜孔的聚乙二醇无法抑制紫杉烷或 5‑FU 诱导的 HMGB1 减少。死活区分染料染色清晰显示,DTX 或 FdUMP 处理的完整活细胞内 HMGB1 显著降低(图 2A)。
HMGB1 无分泌信号肽,无法通过经典内质网‑高尔基体途径分泌。已知单核细胞与巨噬细胞在脂多糖(LPS)等炎症刺激下,可通过核输出蛋白 CRM1 将 HMGB1 从细胞核主动转运至胞质并分泌。如图 2B 所示,CRM1 抑制剂细霉素 B 可完全阻断 DTX 或 PTX 诱导的 PyMT‑B6 细胞核内 HMGB1 丢失,但对 FdUMP 诱导的 HMGB1 丢失无效。
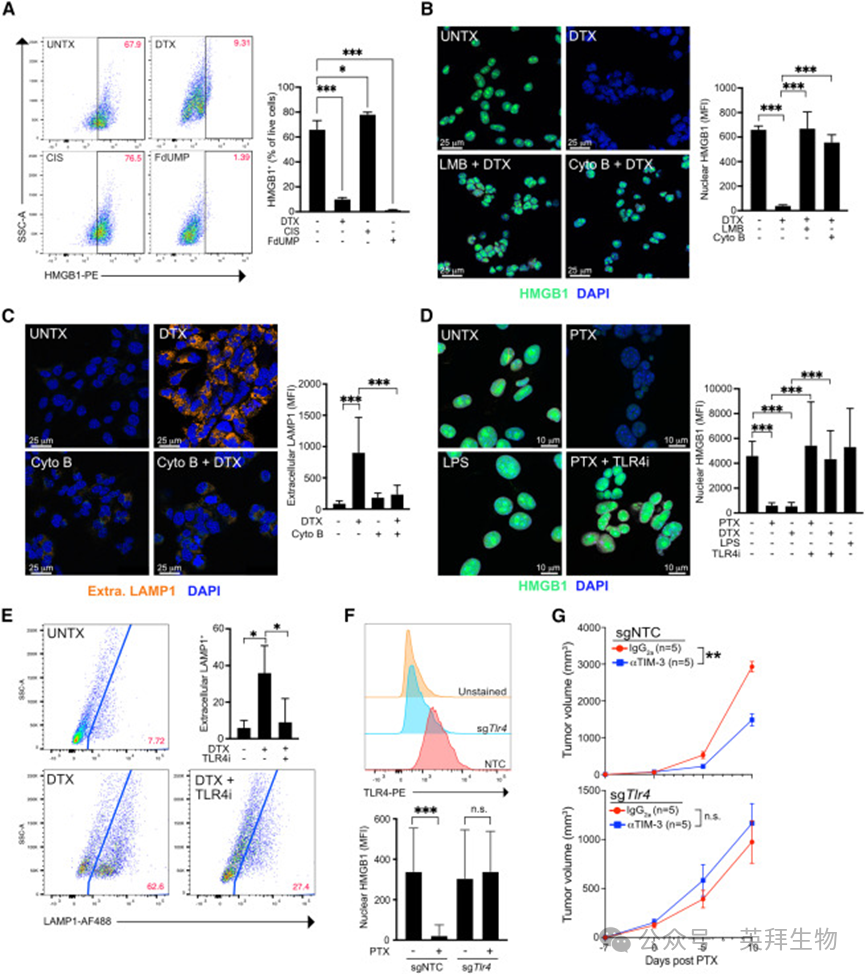
图2. 紫杉烷治疗期间 HMGB1 的主动分泌依赖于TLR4
巨噬细胞中,胞质 HMGB1会被装入 LAMP1⁺溶酶体囊泡,通过胞吐作用释放。本研究中,紫杉烷处理的肿瘤细胞表面LAMP1 染色显著增加(图 2C);肌动蛋白抑制剂细胞松弛素 B 可阻断 LAMP1 表面表达,同时抑制 HMGB1 释放(图 2B)。有趣的是,尽管 FdUMP 诱导的 HMGB1 释放不依赖 CRM1 核输出,仍可被细胞松弛素 B 抑制。因此,紫杉烷与 5‑FU 均诱导 HMGB1主动分泌,其中紫杉烷的分泌模式与巨噬细胞相似。
巨噬细胞中 TLR4 信号足以驱动 HMGB1 主动分泌;紫杉烷是 TLR4 及其辅助蛋白 MD‑2 的配体,且 PTX 可诱导小鼠巨噬细胞主动分泌 HMGB1。流式检测显示,小鼠 PyMT‑B6、KP1、4T1、E0771 细胞系均高表达 TLR4,且均可高效分泌 HMGB1;人 MDA‑MB‑231 与 MCF7 细胞同样高表达 TLR4,与既往报道一致。
TLR4 药理学抑制剂 M62812 可阻断 HMGB1 释放(图 2D、S2G)与胞吐作用(图 2E),且单独使用不影响 HMGB1 定位。CRISPR/Cas9 构建的 Tlr4 敲除 PyMT‑B6 细胞在 PTX 处理后完全不释放 HMGB1(图 2F)。
最后我们评估 Tlr4 敲除肿瘤对 TIM‑3 阻断的响应。PyMT‑B6 细胞较原代 PyMT 肿瘤侵袭性更强,Tlr4 敲除可减缓其生长,与既往研究一致(35,36)(图 2G)。关键发现:Tlr4 敲除肿瘤对 PTX 联合 TIM‑3 阻断完全无响应。该结果支持 TLR4 信号驱动 HMGB1 释放,但也不能排除化疗疗效因肿瘤生长减慢而降低的可能。与巨噬细胞HMGB1 分泌不同,LPS 单独刺激无法诱导肿瘤细胞释放 HMGB1(图 2D),提示 TLR4 激活是必要非充分条件,紫杉烷需激活额外通路。
紫杉烷诱导 ROS 依赖性 DNA 损伤与 PARP 激活
巨噬细胞中,TLR4 激活产生活性氧(ROS),使 HMGB1 的 A 盒结构域形成二硫键(C23‑C45),干扰核定位信号、削弱 DNA 结合能力,进而促进与CRM1 结合。本研究中,DTX 处理后细胞脂质过氧化与氧化活性显著升高,间接提示 ROS 水平上升(图 3A)。
既往研究显示,巨噬细胞中TLR4 诱导的脂质过氧化可被TLR4 抑制剂或 NADPH 氧化酶(NOX)抑制剂逆转,从而减少 ROS 生成。本研究中,NOX 抑制剂或 ROS 清除剂 N‑乙酰半胱氨酸(NAC)可降低 DTX 诱导的 ROS 水平,并阻断 HMGB1 释放(图 3A、3B);抑制剂单独使用不影响 HMGB1 含量。与之不同,FdUMP 诱导的 HMGB1 释放不依赖 ROS,符合其 CRM1 非依赖的核输出途径。
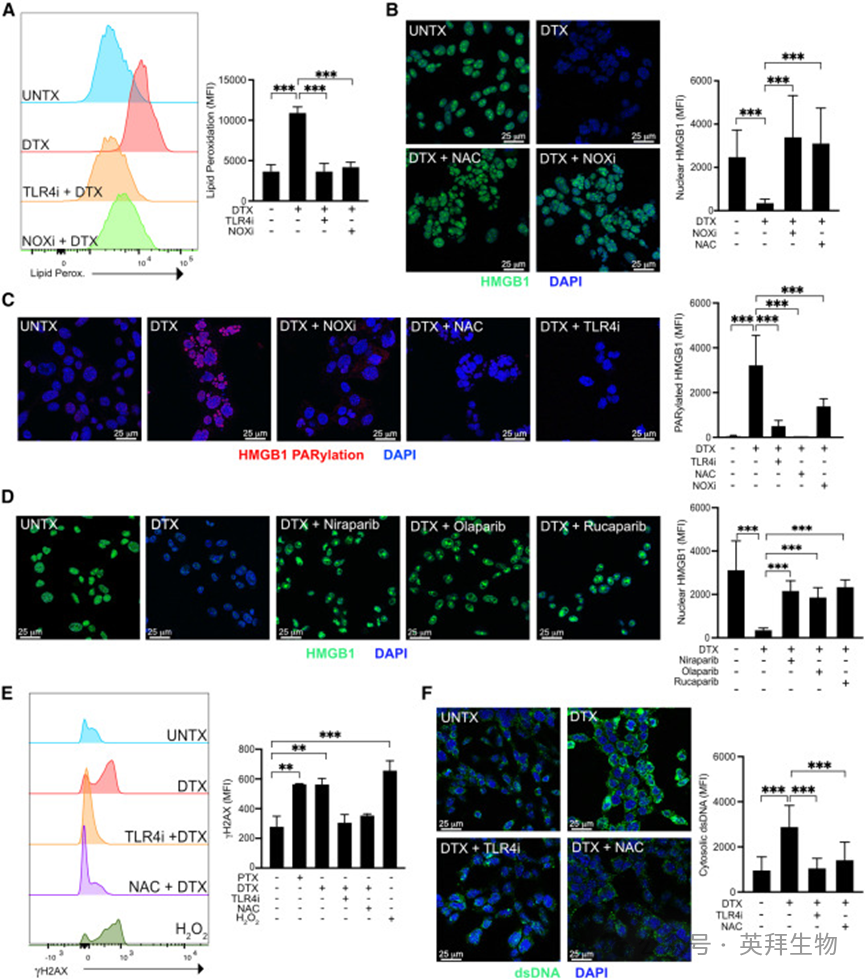
图3. 紫杉烷诱导 ROS 依赖性DNA 损伤与 PARP 激活
巨噬细胞中 HMGB1 核输出的另一关键翻译后修饰是 PARP1 介导的多聚 ADP 核糖化(PARylation),该过程由 DNA 链断裂激活。本研究中,DTX 处理后细胞总 PARylation 水平升高,组蛋白乙酰转移酶活性因 PARP1 激活而上调;邻近连接实验(PLA)直接检测到 HMGB1 的 PARylation 特异性增加(图 3C),且该修饰可被 TLR4 抑制剂或 NAC 阻断(图 3C)。PARP1/2 抑制剂(尼拉帕利、奥拉帕利、卢卡帕利)均可阻断 HMGB1 释放(图 3D)。以上结果提示,紫杉烷诱导的 DNA 断裂激活 PARP1,促使 HMGB1 从细胞核转位至胞质。
紫杉烷虽不直接损伤 DNA,但可导致子代细胞持续多极性分裂,增加细胞分裂时基因组改变风险,引发染色体不稳定性,最终导致双链 DNA(dsDNA)断裂,这一现象在 MMTV‑PyMT 肿瘤细胞中已被。我们推测该 DNA 损伤源于 TLR4 阳性细胞的高氧化应激(图 3A),联合紫杉烷导致的多极性纺锤体形成引发核膜破坏。结果显示,PTX 或 DTX 处理后磷酸化 H2AX(γH2AX)显著升高,提示 dsDNA 断裂(图 3E);DTX 处理细胞中剪切型 PARP1 的出现进一步证实 DNA 损伤反应与 PARP 激活。同时,胞质 dsDNA 含量显著增加(图 3F);TLR4 抑制剂或 NAC 可逆转 γH2AX 升高与胞质 dsDNA 积累(图 3E、3F)。综上,ROS 依赖性 DNA 损伤激活 PARP1,HMGB1 发生 PARylation 后从细胞核转位至胞质。
cGAS‑STING通路是 HMGB1 释放的必需条件
基于紫杉烷诱导基因组不稳定性与胞质 dsDNA 积累的结果,我们探究 cGAS‑STING 通路是否参与 HMGB1 释放。使用已构建的 Cgas 与 Sting 敲除 PyMT‑B6 细胞,发现任一基因缺失均可完全阻断 DTX 诱导的 HMGB1 释放(图 4A)。
将敲除细胞原位接种后评估PTX 联合 TIM‑3 阻断的疗效,结果与体外一致:肿瘤细胞 Cgas 或 Sting 敲除可完全消除联合治疗的增效作用(图 4B)。与 TLR4 敲除不同,STING 缺失不改变肿瘤生长速率或对 PTX 的单药敏感性,也不影响 PTX 体内处理后 CD45⁻肿瘤细胞中 γH2AX 水平。对照组、sgTlr4 与 sgSting 肿瘤的 Ki67 增殖染色无差异,提示上述基因不通过改变细胞周期影响肿瘤对 PTX 的敏感性。该结果支持 STING 在 DNA 损伤与胞质 dsDNA 积累下游调控 HMGB1 释放。
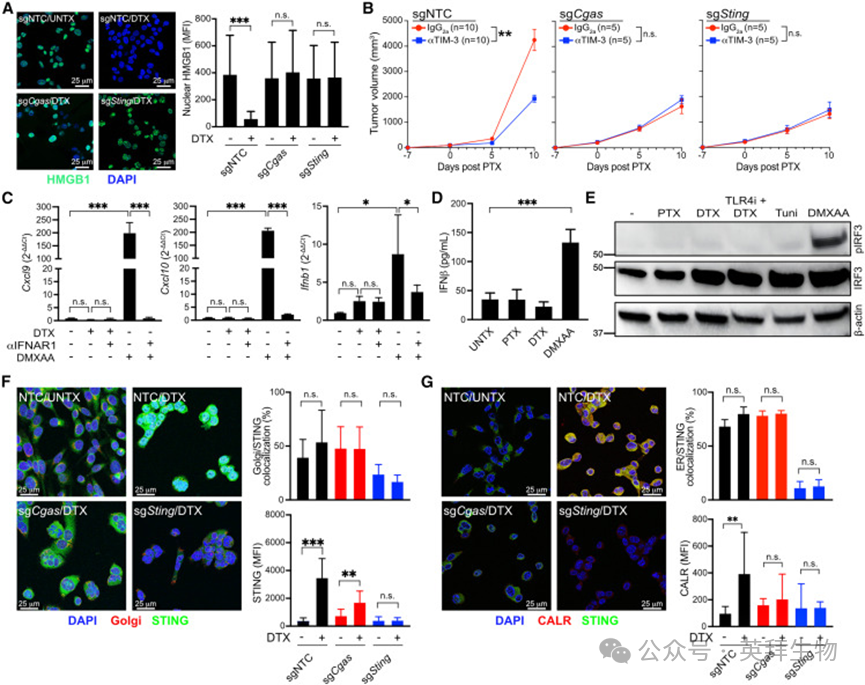
图4. cGAS-STING 通路是 HMGB1 释放所必需的
经典 STING 激活通路中,cGAS 识别胞质 DNA 产生 2′3′‑cGAMP,促使 STING 从内质网转位至高尔基体,进而磷酸化 TBK1 与 IRF3,诱导 Ⅰ 型干扰素(IFN)表达。但本研究中,DTX 处理未上调 Ifnb1 mRNA,也未激活 IFN 应答基因 Cxcl9 与 Cxcl10(图 4C);与之对比,STING 激动剂 DMXAA 可强烈诱导上述基因表达。DTX 与 PTX 同样无法上调上清中 IFN‑β 含量(图 4D),且未检测到 IRF3 磷酸化(图 4E)。Ⅰ 型干扰素受体(IFNAR1)阻断不影响紫杉烷诱导的HMGB1 释放或 LAMP1 表面表达(溶酶体胞吐标志物)。
高尔基体转位是 STING 激活 TBK1 与诱导 Ⅰ 型 IFN 的必需步骤,因此我们分析DTX 处理后 STING 的定位。结果显示,STING 未发生高尔基体转位,但 DTX 处理后细胞内总 STING 蛋白显著增加(图 4F);cGAS 敲除可大幅逆转该效应,提示胞质 dsDNA 积累下游产生的 2′3′‑cGAMP 驱动 STING 蛋白上调(图 3F)。我们进一步用 STING‑ZsGreen 融合蛋白细胞验证:DTX 处理后 STING 不移位至高尔基体,而 DMXAA 可诱导该转位。共染内质网标志物钙网蛋白显示,DTX 处理后 STING 与内质网高度共定位(图 4G、S4G);同时钙网蛋白染色显著增强,提示内质网扩张,这是未折叠蛋白反应(UPR)的典型特征。上述扩张在Cgas 与 Sting 敲除细胞中消失,证明cGAS‑STING 通路介导内质网扩张。综上,紫杉烷激活的 STING 将基因组不稳定性与胞质 DNA 感知传导至内质网应激通路。
紫杉烷诱导 STING 依赖性内质网应激与 HMGB1 胞吐
为直接验证紫杉烷是否诱导内质网应激,我们用内质网荧光探针(ER Tracker Blue‑White)定量细胞器扩张。结果显示,DTX 诱导的内质网扩张程度与经典内质网应激诱导剂衣霉素相当(图 5A);NAC、NOX 抑制剂或 TLR4 抑制剂可逆转该扩张,提示内质网应激由 ROS 依赖性 DNA 损伤与 cGAS‑STING 激活驱动。Cgas 或 Sting 敲除细胞在 DTX 处理后完全不发生内质网应激,且对衣霉素诱导的内质网应激也表现抵抗,与高染色体不稳定性乳腺癌细胞的近期报道一致。
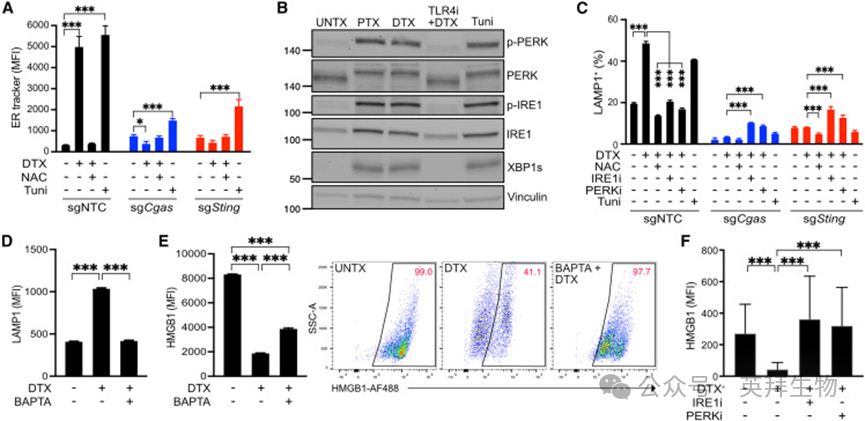
图5. 紫杉烷诱导 STING 依赖性内质网应激与HMGB1 胞吐作用
我们通过检测 IRE1α 与 PERK 磷酸化、XBP1 剪切体(XBP1s,UPR 核心转录因子)证实 DTX 激活 UPR(图 5B)。尽管 cGAS‑STING 对内质网应激至关重要,其缺失不改变 PyMT‑B6 细胞对紫杉烷的体外敏感性。
为明确内质网应激与 UPR 对 HMGB1 释放的功能作用,我们首先分析其对溶酶体胞吐的影响:该过程依赖胞质钙流,而内质网是细胞内主要钙库。衣霉素可诱导 LAMP1 表面表达,程度与 DTX 相当(图 5C),提示内质网应激足以驱动胞吐;但单纯内质网应激无法诱导 HMGB1 释放,可能因衣霉素直接诱导应激时缺乏 HMGB1 核转位的前置步骤。
钙螯合剂 5,5′‑二甲基 BAPTA 可阻断 DTX 诱导的溶酶体胞吐(图 5D)与 HMGB1 释放(图 5E),但无法将胞内 HMGB1 完全恢复至未处理水平,原因可能是 HMGB1 在溶酶体中降解或钙螯合的其他效应。最后,PERK 或 IRE1α 抑制剂可阻断 DTX 诱导的胞吐(图 5C)与 HMGB1 释放(图 5F),单独使用抑制剂无效应。综上,紫杉烷通过非经典 STING 通路触发内质网应激、UPR 激活与钙依赖性溶酶体胞吐,这些步骤均为 HMGB1 释放所必需。
STING是三阴性乳腺癌化疗免疫治疗的必需因子
我们前期已证明转基因 C3 (1)‑TAg TNBC 模型对 PTX 联合抗 TIM‑3 治疗的响应与 MMTV‑PyMT 模型相似,且人 MDA‑MB‑231 细胞呈现 TLR4 依赖性 HMGB1 释放。因此我们在 KrasG12D/Trp53 缺失驱动的 TNBC 细胞系 KP1 中验证结论,该细胞 TLR4 表达水平与 MDA‑MB‑231 相近。
与前期结果一致,KP1 细胞以TLR4依赖方式释放 HMGB1,且紫杉烷处理后胞质 dsDNA 增加(图 6A、6B);DTX 同样以 TLR4 与 ROS 依赖方式诱导内质网应激与溶酶体胞吐(图 6C、6D)。关键动物实验显示:原位 KP1 肿瘤对 PTX 联合抗 TIM‑3 治疗响应良好,而 Sting 敲除肿瘤完全无效(图 6E)。该效应并非源于经典STING 激活:紫杉烷无法诱导STING 磷酸化或 Cxcl9 表达,而 DMXAA 可实现。
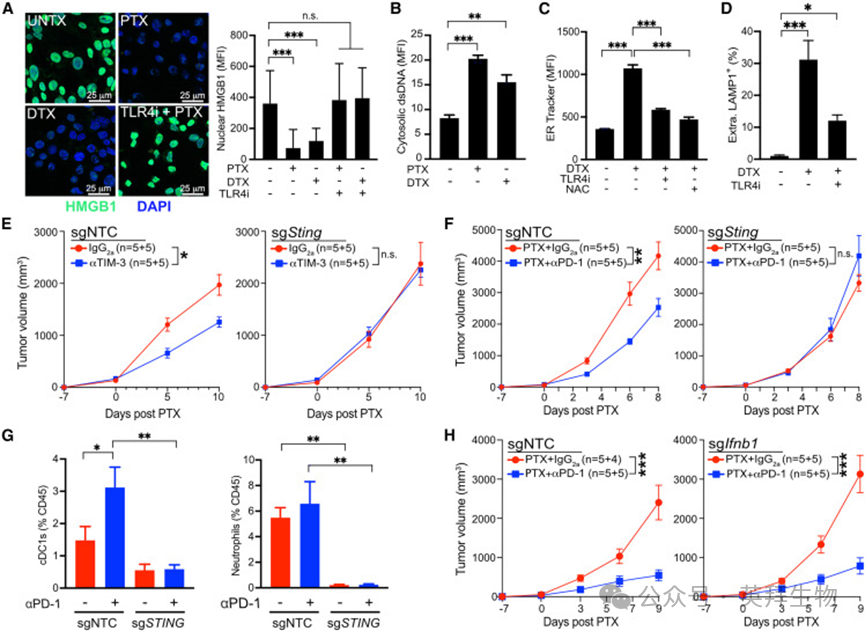
图6. 三阴性乳腺癌模型中 STING 依赖性内质网应激与HMGB1 释放
为探索 HMGB1 释放与乳腺癌患者预后的关联,我们构建 HMGB1 释放基因特征(PARP1、KAT2B、CREBBP、EP300、IRF1、XPO1),聚焦 HMGB1 乙酰化与 PARylation 相关的翻译后修饰及下游通路。该特征与乳腺癌患者(尤其 TNBC)的 T 细胞浸润呈正相关,且与 TNBC 患者无进展生存期显著改善相关。
抗 PD‑1(帕博利珠单抗)联合化疗已成为高危早期或晚期 TNBC 的标准治疗,本研究中 KP1 模型对 PTX 联合抗 PD‑1 治疗响应良好。鉴于紫杉烷无法诱导 Ⅰ 型 IFN 反应,我们探究 STING 在该化疗免疫方案中的作用。结果显示:Sting 野生型肿瘤对抗 PD‑1 联合 PTX 显著响应,Sting 敲除肿瘤则完全无效(图 6F)。该差异并非由 MHCI 或 PD‑L1 表达改变导致:STING 缺失在体外与体内均不影响上述分子表达,符合 STING 非依赖 Ⅰ 型 IFN 的结论。
对 sgNTC 与 sgSting 肿瘤的流式分析显示,PTX 处理后 Sting 敲除肿瘤招募 CD103⁺ cDC1 与 Ly6G⁺中性粒细胞的能力下降,F4/80⁺巨噬细胞比例相对上升,淋巴细胞亚群无显著差异(图 6G)。为明确 Ⅰ 型 IFN 的作用,我们构建 Ifnb1 敲除 KP1 肿瘤,结果显示 sgNTC 与 sgIfnb1 肿瘤对 PTX 联合抗 PD‑1 治疗均有效(图 6H)。
综上,肿瘤细胞固有STING 是紫杉烷治疗中髓系细胞浸润的关键调控因子,且以 Ⅰ 型 IFN 非依赖方式决定 TNBC 抗 PD‑1 化疗免疫治疗的疗效,其具体机制可能为内质网应激、胞吐作用或其他分子通路,有待进一步研究。
参考文献
Onimus A, Celias D, Chang S, Davis J, Mandula J, Li J, Gardner A, Hänggi K, Osunmakinde O, Soliman H, Shaw TI, Rodriguez PC, Ruffell B. Taxane chemotherapy promotes response to TIM-3 checkpoint blockade via STING-mediated ER stress and HMGB1 secretion. Cell Rep Med. 2026 May 6:102788. doi: 10.1016/j.xcrm.2026.102788. Epub ahead of print. PMID: 42097146.