






SPOP 的 O-GlcNAc 糖基化通过介导β-连环蛋白降解来调节结直肠癌进展和铁死亡
目前结直肠癌(CRC)的治疗手段面临复发和耐药等挑战。铁死亡是一种新型的细胞死亡形式,是治疗结直肠癌的有前景的方法。SPOP 作为 E3 泛素连接酶复合物 CRL3 的底物结合蛋白,在生物学中发挥着重要作用,但其在结直肠癌患者中的治疗效果以及对铁死亡的调节能力仍知之甚少。本研究证明 SPOP 在结直肠癌中发挥着肿瘤抑制作用,能够抑制结直肠癌细胞的增殖和转移,并提高其对铁死亡的敏感性。转录组分析表明 Wnt 信号通路可能是 SPOP 功能的一个潜在靶点。进一步的数据表明,SPOP 敲低会增加β-连环蛋白的蛋白水平,而临床数据则表明 SPOP 表达对β-连环蛋白的蛋白水平有相反的影响。分子生物学实验表明 SPOP 促进β-连环蛋白 K508 位点的多泛素化和降解。有趣的是,SPOP 的 O-GlcNAc 糖基化会降低其蛋白质稳定性,并影响 SPOP 与β-连环蛋白的结合,SPOP 还通过抑制β-连环蛋白/SLC7A11 轴促进结直肠癌细胞的铁死亡。SPOP 靶向药物马普替林与铁死亡诱导剂联合治疗在结直肠癌细胞和异种移植瘤中具有协同抗肿瘤效果。我们的研究揭示了 SPOP 在结直肠癌中的多方面功能,激活 SPOP 可能是提高结直肠癌对铁死亡诱导剂敏感性的一种可行策略。本文于2025年11月发表于Cell Death Discovery(IF=7)上。
技术路线:

结果:
1)SPOP在结直肠癌中表达水平较低,且与较长的预后相关
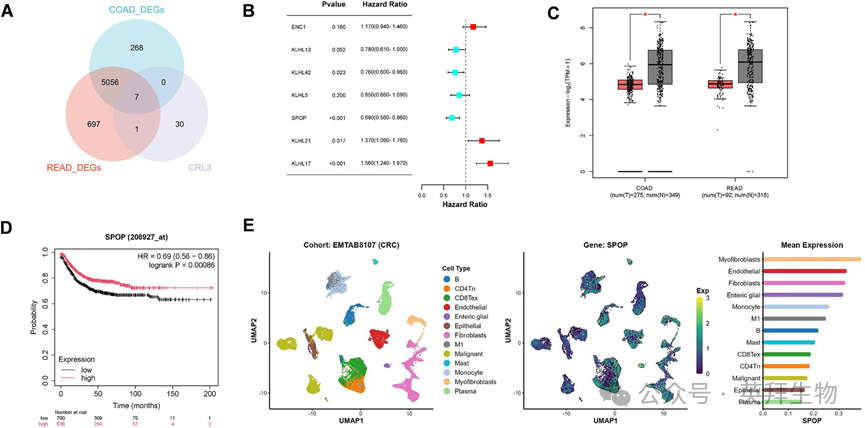
我们首先从IUUCD数据库中收集到CRL3相关基因列表(n = 37),然后通过TCGA-COAD和TCGA-READ获得差异表达基因(DEGs),它们相交得到7个候选基因(图1A)。结合这7个基因在CRC中的预后,我们筛选出候选基因SPOP(图1B)。通过在线数据库,我们发现SPOP在结直肠癌组织中的表达水平低于正常组织(图1C),并且与结直肠癌患者更长的生存期相关(图1D)。为了进一步探讨SPOP在CRC亚群中的表达,我们分析了CRC单细胞数据集(EMTAB8107),结果显示SPOP的表达主要集中在肌成纤维细胞中,而在肿瘤细胞亚群中的表达相对较低(图1E)。这些发现表明,SPOP在结直肠癌中表达下调,并与较长的预后相关。
2)SPOP抑制结直肠癌进展
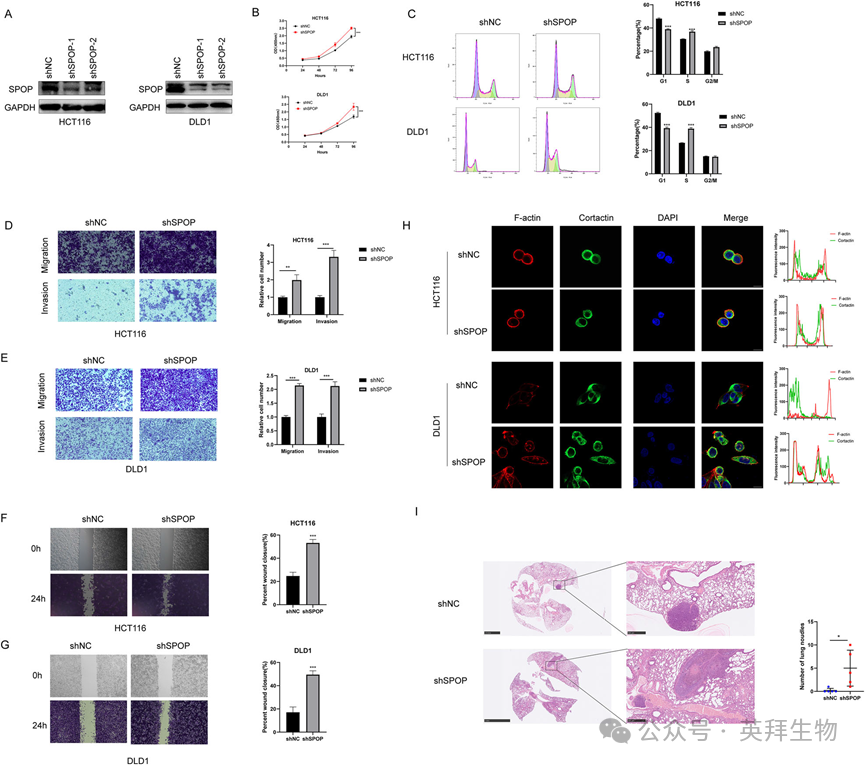
为了验证SPOP在CRC中的生物学功能,我们首先构建了SPOP敲低细胞系(图2A),并通过CCK8实验发现SPOP敲低增加了CRC细胞的增殖能力(图2B),而周期实验表明SPOP可以改变CRC细胞的周期分布(图2C)。我们还通过伤口愈合和transwell实验发现,SPOP的下调促进了结直肠癌细胞系的迁移和侵袭(图2D-G)。IF显示在敲除SPOP后,Cortactin和F-actin的共定位明显增加(图2H),提示Invadopodia的增加。然后我们构建了肺转移定植模型来验证SPOP对转移的影响,结果显示shSPOP组有更多的转移灶(图2I),表明SPOP可以抑制转移。这些结果表明SPOP在结直肠癌中具有肿瘤抑制作用。
3)SPOP与β-catenin结合
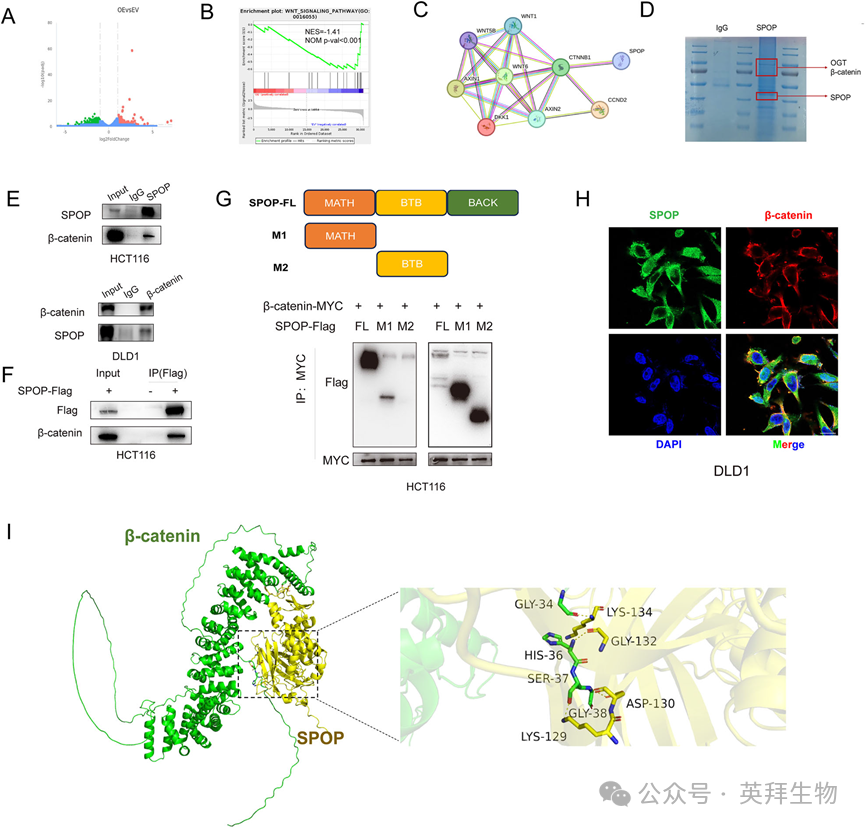
为了探索SPOP在结直肠癌中的作用机制,我们对SPOP过表达的HCT116细胞进行了RNA测序(图3A)。基于RNA-seq的GSEA结果表明,SPOP过表达组的Wnt信号通路活性降低(图3B)。接下来,我们通过STRING数据库分析Wnt信号通路相关分子的PPIs,发现SPOP与β-catenin结合(图3C)。此外,我们通过IP-MS鉴定了SPOP结合蛋白(图3D)。我们通过co-IP证实了SPOP和β-catenin之间存在内源性和外源性相互作用(图3E,F)。SPOP作为E3连接酶的作用依赖于MATH结构域。Co-IP结果显示,具有MATH结构域的截断突变体可以与β-catenin结合,而具有BTB结构域的截断突变体不能与β-catenin结合(图3G)。免疫荧光进一步证实了SPOP与β-catenin在结直肠癌细胞中的共定位(图3H)。此外,分子对接模型预测了SPOP结构域中与β-catenin结合的特定残基(图3I)。这些结果表明SPOP的Lys129、Asp130、Gly132和Lys134残基是与β-catenin结合的关键残基。据报道,SPOP的G132V突变是一种致病性错义突变,可增加SPOP的已知底物BRD4的表达水平。因此,我们构建了SPOPG132V点突变质粒,Co-IP结果显示SPOP-G132V突变减少了与β-catenin的结合,提示G132位点是SPOP与β-catenin的结合位点。
4)SPOP调控β-catenin的表达
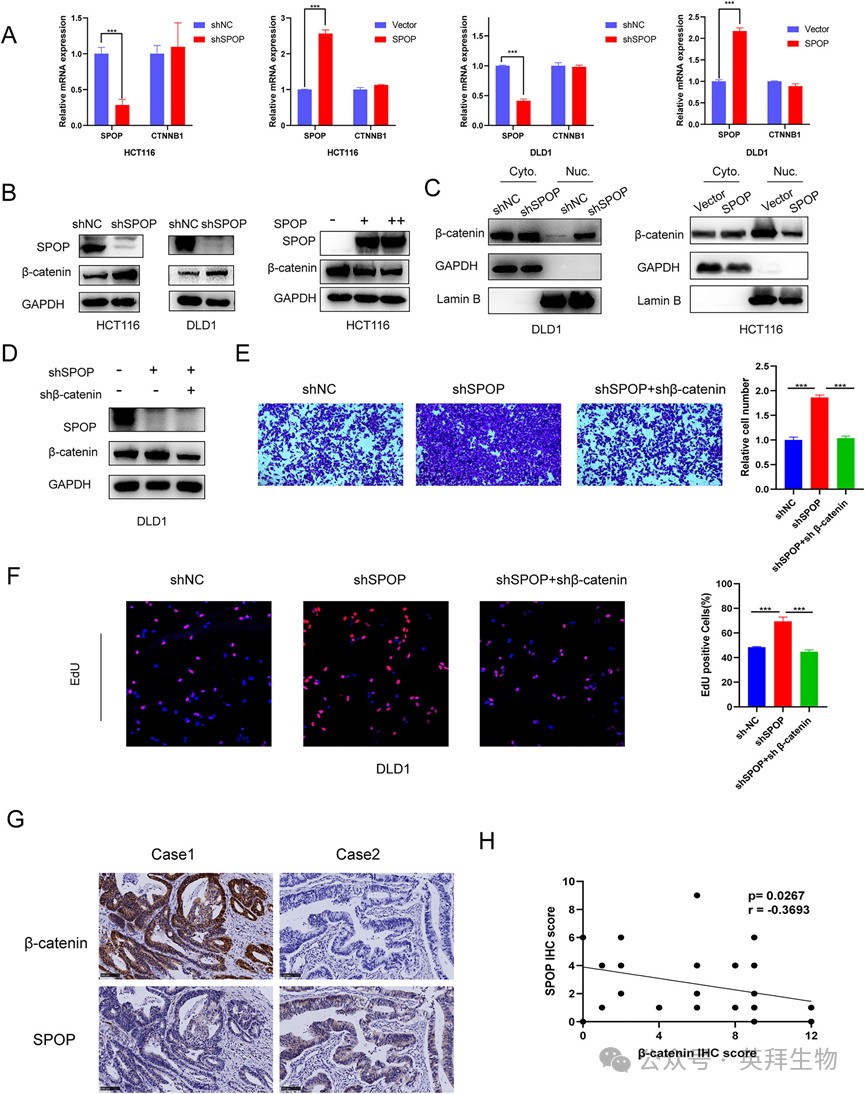
在结直肠癌细胞中,SPOP的过表达和敲低均不影响β-catenin的mRNA水平(图4A)。然而,敲低SPOP增加了β-catenin蛋白水平,而过表达SPOP则降低了β-catenin蛋白水平(图4B),这表明SPOP可能通过翻译后修饰(PTM)途径调节β-catenin。我们证实了细胞核β-catenin在SPOP被敲低后细胞中增加,而在SPOP过表达后细胞中减少(图4C)。为了验证SPOP和β-catenin在结直肠癌中的联合作用,我们构建了共转染细胞系(图4D)。transwell和EdU实验显示,SPOP和β-catenin的联合敲低减弱了SPOP敲低对CRC细胞的促侵袭和增殖作用(图4E,F)。通过免疫组化分析CRC组织中SPOP和β-catenin的蛋白表达水平。结果表明,CRC患者组织中SPOP和β-catenin丰度之间存在显著的负相关(图4G,H)。
5)SPOP促进β-catenin的泛素化
考虑到SPOP作为E3泛素连接酶,基于前期实验结果,我们提出SPOP可能通过泛素-蛋白酶体系统下调β-catenin蛋白水平。为了检验这种可能性,我们用蛋白酶体抑制剂MG132处理过表达SPOP的HCT116和DLD1细胞。结果显示,MG132可以抵消SPOP表达升高引发的β-catenin降解(图5A)。此外,使用环己亚胺(CHX)抑制蛋白质合成,我们观察到HCT116细胞中SPOP敲低导致β-catenin半衰期延长(图5B),表明其在蛋白质周转中的作用。泛素化实验进一步支持了这种调节作用:沉默SPOP导致β-catenin泛素化减少(图5C),而过表达SPOP则增加了其泛素化水平(图5D)。由于SPOP的MATH结构域与β-catenin结合,并且MATH结构域识别底物蛋白的SBC基序,因此我们推断了β-catenin的两个SBC基序,并构建了两个β-catenin截断质粒(SBC1和SBC2)(图5E)。Co-IP发现β-catenin-SBC2突变体失去了与SPOP结合的能力(图5F),并且β-catenin-SBC2突变体的泛素化水平显著降低(图5G)。过表达SPOP显著增加了β-catenin的K27连锁多泛素化,但不影响K48和K63连锁的β-catenin的多泛素化(图5H)。为了确定SPOP识别的β-catenin上的赖氨酸位点,我们通过PhosphoSitePlus®数据库预测了β-catenin的赖氨酸位点,选择了前3个赖氨酸残基(K170、K233和K508),并构建了针对这些赖氨酸残基的突变体来验证我们的假设。我们发现只有K508R突变阻断了SPOP介导的β-连环蛋白泛素化(图 5I)。综上所述,SPOP促进了K508残基上β-连环蛋白的泛素化。
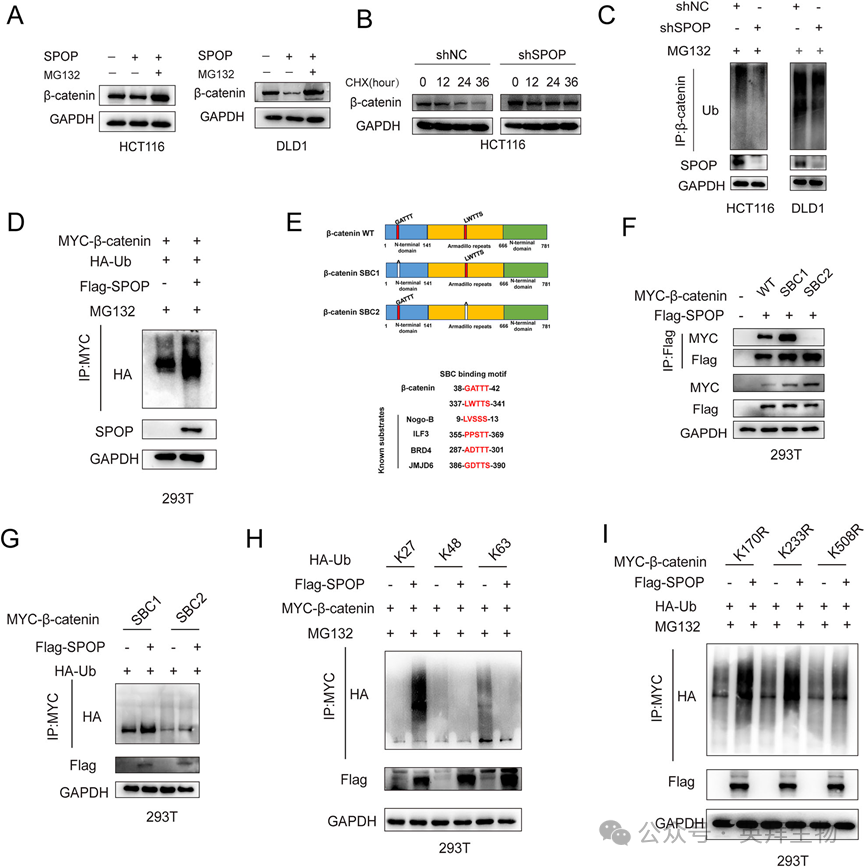
6)SPOP的O-GlcNAcylation影响SPOP与β-catenin的结合
IP-MS结果表明,OGT是SPOP的结合蛋白,由于Zhou等人报道了SPOP和OGT可以结合PDAC和293 T细胞系,我们通过Co-IP验证了这一点,发现SPOP和OGT也可以结合CRC细胞系(图6A)。OGT过表达时,O-GlcNAc水平升高,而SPOP蛋白表达水平降低(图6B),OGT敲低后,结果与OGT过表达相反(图6C)。为了进一步验证OGlcNAc对SPOP表达的调节作用,在OGT抑制剂OSMI-1处理后,SPOP水平上调(图6D),而在OGA(去除O-GlcNAc的酶)抑制剂TMG处理后,HCT116细胞中O-GlcNAc水平上调,但SPOP蛋白水平下调(图6E)。这些结果表明,结直肠癌中SPOP蛋白水平受O-GlcNAc水平的负调控。接下来,我们试图探索泛素-蛋白酶体途径是否参与OGT介导的SPOP降解。MG132处理过表达OGT的HCT116细胞。结果显示,MG132处理阻断了OGT过表达引起的SPOP降解(图6F),并且OGT过表达增加了SPOP的泛素化(图6G),这表明O-GlcNAc通过蛋白酶体依赖性途径调节SPOP降解。接下来,我们使用质谱法鉴定HCT116细胞SPOP上的O-GlcNAc位点。我们确定了三个可能的糖基化位点(S58、S59和T114)(图6H)。在这些结果的基础上,我们生成了不同的SPOP点突变体(S58/S9A和T114A),并检测了它们的O-GlcNAc水平,发现S58/S9A突变体的O-GlcNAc结合明显降低(图6I),提示S58/S9A可能是SPOP上的O-GlcNAc位点。为了进一步证实SPOP相对于O-GlcNAc的稳定性,我们检测了SPOP突变体S58/59A的半衰期,该突变体的降解率高于SPOP-WT(图6J),并且SPOP突变体S58/59A的泛素化水平高于SPOP-WT(图6K)。我们证明SPOP与β-catenin结合并促进其降解,由此推测SPOP的O-GlcNAcylation可能影响其与β-catenin的关系。接下来,我们发现SPOP突变体S58/59 A的β-catenin蛋白水平低于SPOP-WT,SPOP突变体S58/59A与β-catenin的结合明显大于SPOP-WT组(图6L,M),β-catenin泛素化可以增加(图6N)。综上所述,这些结果表明SPOP的O-GlcNAcylation通过抑制其与β-catenin的结合从而抑制β-catenin的降解,从而促进β-catenin的表达。
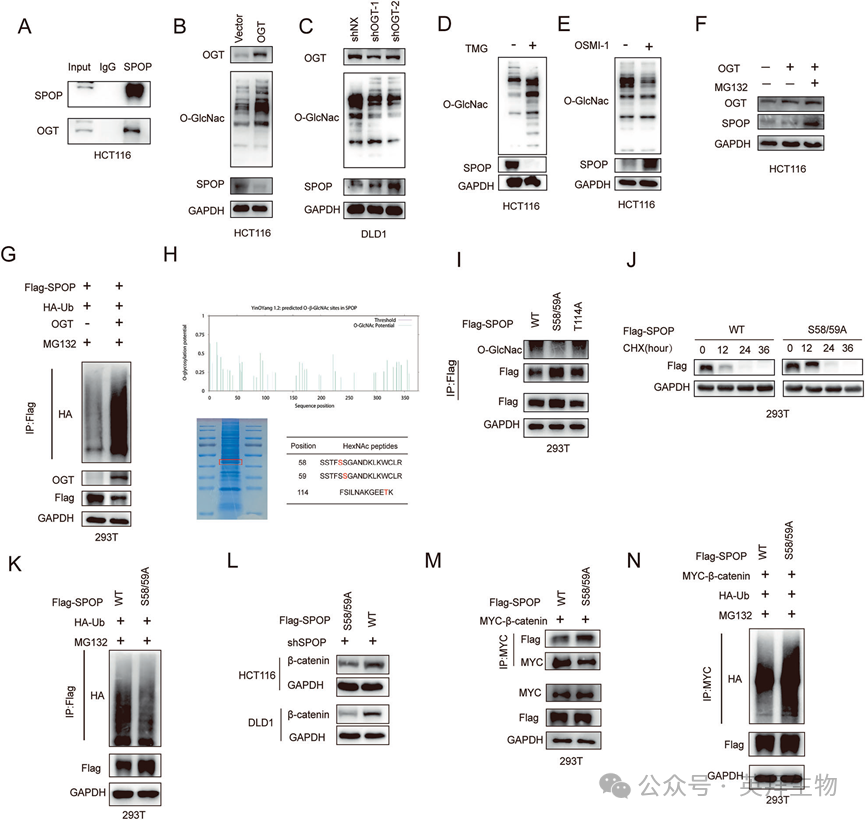
7)SPOP诱导结直肠癌铁死亡
为了进一步研究SPOP调控CRC生长的功能,我们对TCGA数据库进行了KEGG分析,结果显示铁死亡位于前10位基因之列(图7A)。因此,我们研究了SPOP与铁死亡之间的关系,我们的结果表明,SPOP过表达显著增强了erastin诱导的对CRC细胞生长的抑制作用。这种抑制作用被铁死亡抑制剂fer1逆转,而其他RCD(坏死性下垂和凋亡)抑制剂,包括Nec-1和Z-VAD-FMK不能逆转(图7B)。我们通过C11-BODIPY和DCFH-DA检测了SPOP对CRC细胞中脂质ROS和ROS水平的影响,发现SPOP的敲低导致CRC细胞中脂质ROS和ROS水平显著降低(图7C)。用FerroOrange探针检测CRC细胞中的Fe2+水平,结果表明,敲除SPOP后,CRC细胞中的Fe2+水平显著降低(图7D)。提示SPOP可诱导结直肠癌细胞对铁死亡的敏感性。
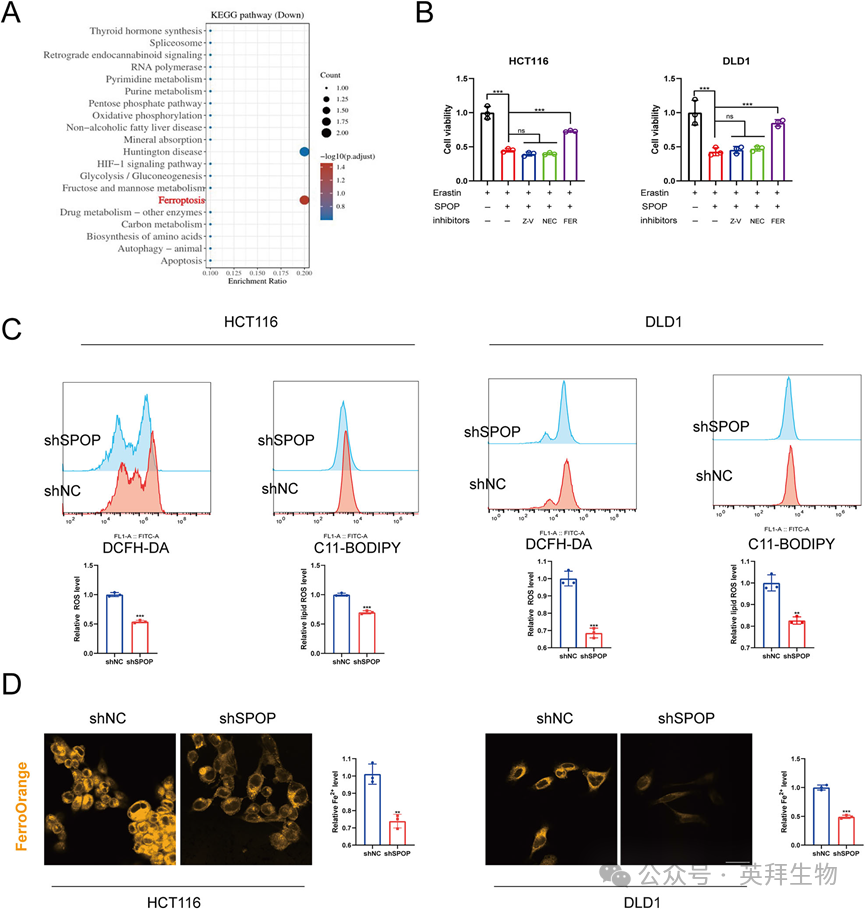
8)SPOP通过调节β-catenin/ SLC7A11轴诱导结直肠癌铁死亡
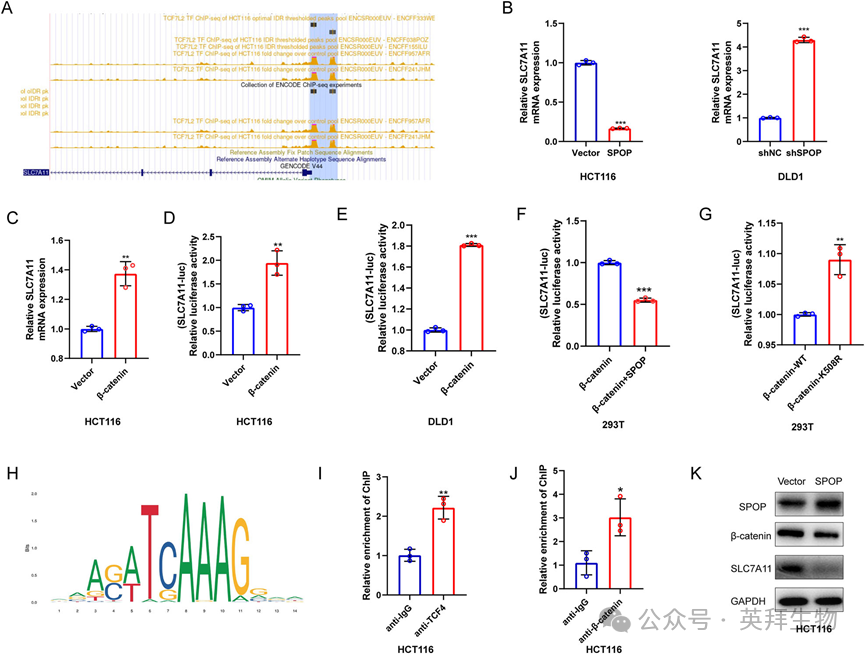
为了阐明SPOP调控CRC细胞铁死亡的机制,我们首先分析了ChIP-Atlas在线数据库。有趣的是,在HCT116细胞系中,SLC7A11的启动子序列出现了β-catenin/TCF4的结合峰(图8A), qPCR提示SPOP可以下调SLC7A11的mRNA水平(图8B),表明SPOP通过转录途径调控SLC7A11。我们之前的研究表明SPOP与β-catenin结合;因此,我们推测SPOP通过β-catenin调控SLC7A11的表达(图8C)。我们首先发现β-catenin的上调增加了SLC7A11的mRNA水平。通过双荧光素酶报告基因检测,我们进一步证实β-catenin增强了SLC7A11启动子的转录活性。相反,SPOP过表达下调了β-catenin上调的SLC7A11启动子活性(图8F),且β-catenin K508R对SLC7A11启动子活性的上调比β-catenin WT对SLC7A11启动子活性的上调更为明显(图8G),提示SPOP可以调节β-catenin对SLC7A11启动子活性的影响。JASPAR数据库预测β-catenin/TCF4与SLC7A11基序结合(图8H);因此,我们通过ChIP-qPCR验证了β-catenin/TCF4与SLC7A11启动子区域的结合(图8I,J),与数据库结果一致。SPOP过表达降低了β-catenin和SLC7A11的蛋白水平(图8K),提示SPOP/ β-catenin/SLC7A11轴可能调节铁死亡。
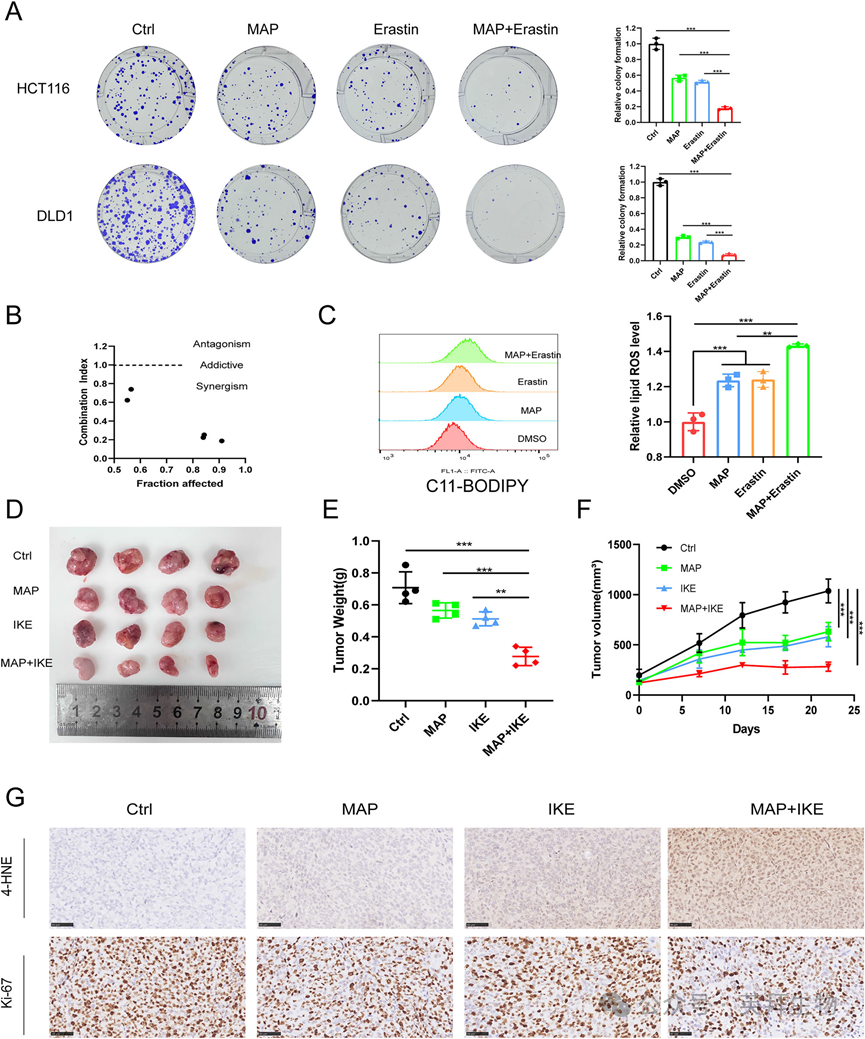
9)马普替林与铁死亡诱导剂IKE具有协同抗癌作用
我们通过MAP与铁死亡诱导剂的结合,探讨MAP是否能增加CRC对铁死亡的敏感性。体外实验显示,与单用相比,MAP与erastin联用可显著抑制结直肠癌细胞的生长(图9A),且MAP与erastin的联合指数小于1(图9B),提示联用具有协同作用。流式细胞术结果还显示,与单独使用Erastin组相比,联合使用组脂质ROS水平显著升高(图9C)。我们通过建立异种移植模型验证联合用药的效果。与单独使用IKE相比,MAP联合IKE可显著降低肿瘤体积和重量,减缓肿瘤生长(图9D-F)。免疫组化染色结果显示,MAP和IKE联合作用可提高肿瘤组织中4-HNE(铁死亡标志物)的表达水平,降低Ki67比值,与前期结果一致(图9G)。综上所述,MAP和IKE联合使用可增强体内对铁死亡的敏感性。
结论:
我们发现了SPOP在CRC中起肿瘤抑制作用,可以增加CRC对铁死亡的易感性。在机制上,SPOP与β-catenin结合并促进其降解和泛素化,从而抑制β-catenin与SLC7A11启动子区域的结合。此外,SPOP可以与O-GlcNAc转移酶OGT相互作用,介导SPOP的O-GlcNAcylation,从而降低SPOP的蛋白稳定性,减弱SPOP与β-catenin的结合,从而促进结直肠癌的进展。我们的研究为结直肠癌治疗提供了新的方向。
参考文献:
Zhang X, Ding Y, Ye Q, Shi S, Zhu N, Weng S, Chen J, Hu W, Yuan Y. O-GlcNAcylation of SPOP regulates colorectal cancer progression and ferroptosis by mediating β-catenin degradation. Cell Death Discov. 2025 Nov 10;11(1):526. doi: 10.1038/s41420-025-02832-y.