






METTL1 介导的 FoxO1 的 m7G 甲基化在代谢功能障碍相关脂肪肝疾病中调节脂质代谢
代谢功能障碍相关脂肪性肝病(MASLD)的特点是肝细胞内脂质积累和变性,其发病机制复杂,使药物开发复杂化。在这项研究中,我们发现METTL1在MASLD小鼠的肝脏和临床样本中上调。肝细胞特异性缺失METTL1抑制脂质合成并促进脂质氧化,减轻高脂肪饮食(HFD)诱导的MASLD小鼠的代谢紊乱。相反,过表达METTL1可以促进脂质合成,同时抑制脂质氧化。m7G测序结果表明,METTL1通过甲基化FoxO1的Exon1区域来调节FoxO1 mRNA的稳定性和蛋白表达水平。此外,我们发现FoxO1的过表达抵消了METTL1缺乏对MASLD小鼠代谢紊乱的保护作用。此外,我们发现了一种有效的METTL1小分子抑制剂,HtMBm,可以显著改善HFD诱导的MASLD。总的来说,我们的研究表明METTL1在MASLD的进展中起着至关重要的作用,并强调了靶向METTL1调节这种情况下脂肪酸代谢的治疗潜力。本文于2025年10月发表于Metabolism(IF=11.9)上。
技术路线:

结果:
1)METTL1表达与脂肪肝疾病相关
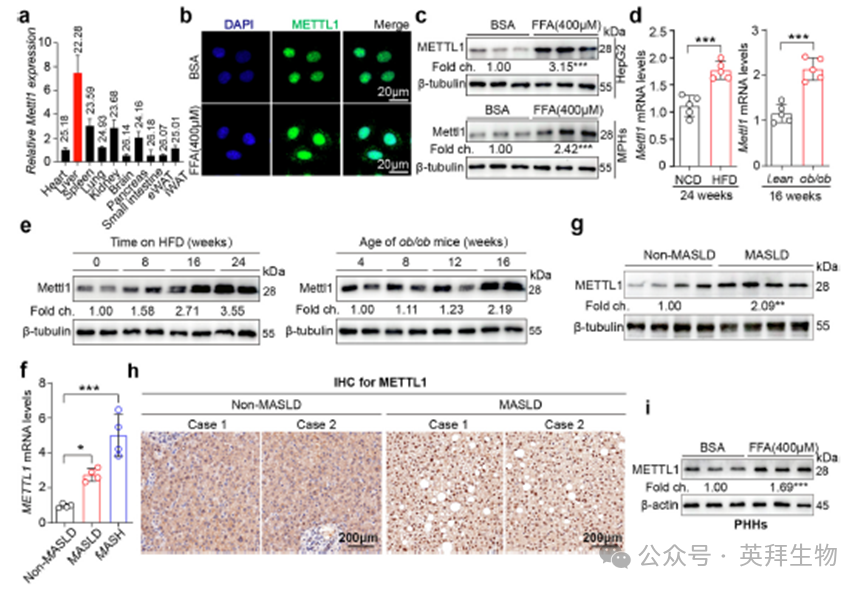
为了研究METTL1与MASLD易感性之间的关系,我们首先检测了METTL1在小鼠不同组织中的差异mRNA表达谱。我们的分析显示,METTL1主要在肝脏中表达,其次是脾脏和肾脏(图1a)。接下来,我们建立了FFAs诱导的肝细胞脂质积累模型,并采用IF和Western blot分析评估METTL1的表达。结果表明,METTL1在这种细胞脂质积累模型中高表达(图1b,c)。此外,通过qPCR和Western blot分析,我们观察到长期HFD治疗小鼠和ob/ob小鼠肝脏中METTL1的表达随着MASLD的进展而稳步增加(图1d,e)。与没有MASLD的个体相比,单纯性脂肪变性或MASH个体肝脏中的METTL1 mRNA水平显著较高(图1f)。值得注意的是,通过免疫组织化学和Western blot分析,与非MASLD组相比,MASLD组个体肝脏细胞核中METTL1的表达明显升高(图1g,h)。此外,我们观察到经FFA处理的原代人肝细胞中METTL1的表达显著增加(图1i)。总的来说,在脂肪肝中观察到的METTL1表达上调表明它可能在MASLD和MASH的进展中发挥重要作用。
2)METTL1缺乏可减轻脂肪肝疾病和代谢紊乱
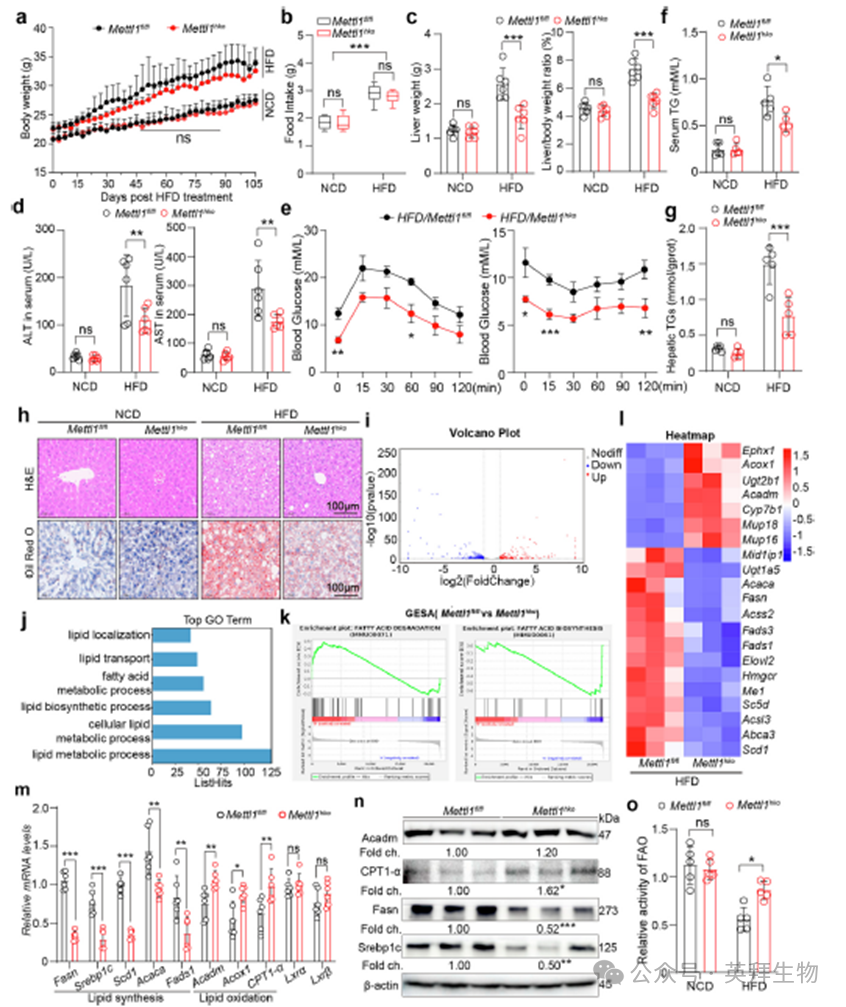
为了研究METTL1对脂肪肝疾病及其并发症的影响,我们培育了肝细胞特异性Mettl1基因敲除小鼠(Mettl1hko),并对它们进行16周的NCD或HFD治疗。在此期间,长期监测显示,尽管与NCD相比,HFD治疗小鼠的体重和食物摄入量明显更高,但Mettl1hko和对照组(Mettl1fl/ fl)小鼠在这些指标上没有显著差异(图2a,b)。然而,在HFD条件下,Mettl1的缺失显著降低了体重和肝体重比,而在NCD条件下则没有(图2c)。此外,ALT和AST水平的测量表明,Mettl1缺乏促进了HFD诱导的MASLD小鼠的肝功能恢复,而不影响NCD条件下的肝功能(图2d)。GTT和ITTs显示,肝细胞中缺乏Mettl1可显著降低胰岛素抵抗(图2e)。组织病理学分析,包括H&E和油红O染色,以及血清和肝脏TG水平的评估显示,与Mettl1fl/fl小鼠相比,Mettl1hko小鼠在HFD应激下表现出显着改善(图2f-h)。为了在转录组水平上系统性地评估Mettl1在代谢功能障碍相关脂肪性肝病(MASLD)发病机制中的作用,我们对HFD喂养的Mettl1hko小鼠和对照Mettl1fl/fl小鼠的肝脏组织进行了RNA-seq分析。差异表达分析显示,与对照组相比,Mettl1hko组中有250个基因上调,286个基因下调(图2i)。GO分析进一步表明,这些差异表达基因主要与脂质代谢相关(图2j)。GSEA发现,在Mettl1hko小鼠中,与脂质代谢相关的通路显著富集(图2k)。转录组分析的热图显示,在HFD喂养的Mettl1hko组小鼠中,与脂质合成相关的基因表达减少(图2l)。此外,qPCR结果证实,在Mettl1缺陷小鼠的肝脏中,脂质合成相关基因(Fasn, Srebp1c, Scd1, Acaca和Fads1)的表达降低,同时参与脂肪酸氧化的基因(Acadm, Acox1和CPT1-α)表达增加(图2m)。Western blot分析进一步验证了Mettl1的缺失抑制了脂质合成,同时促进了脂肪酸氧化(图2n)。此外,FAO实验证实,在HFD条件下,Mettl1缺陷的肝脏表现出增强的脂肪酸氧化活性(图2o)。总之,这些数据表明,Mettl1的缺失通过调控参与脂质合成与氧化的基因表达从而减轻MASLD及其相关的代谢紊乱。
3)METTL1过表达加剧了MASLD小鼠的代谢异常
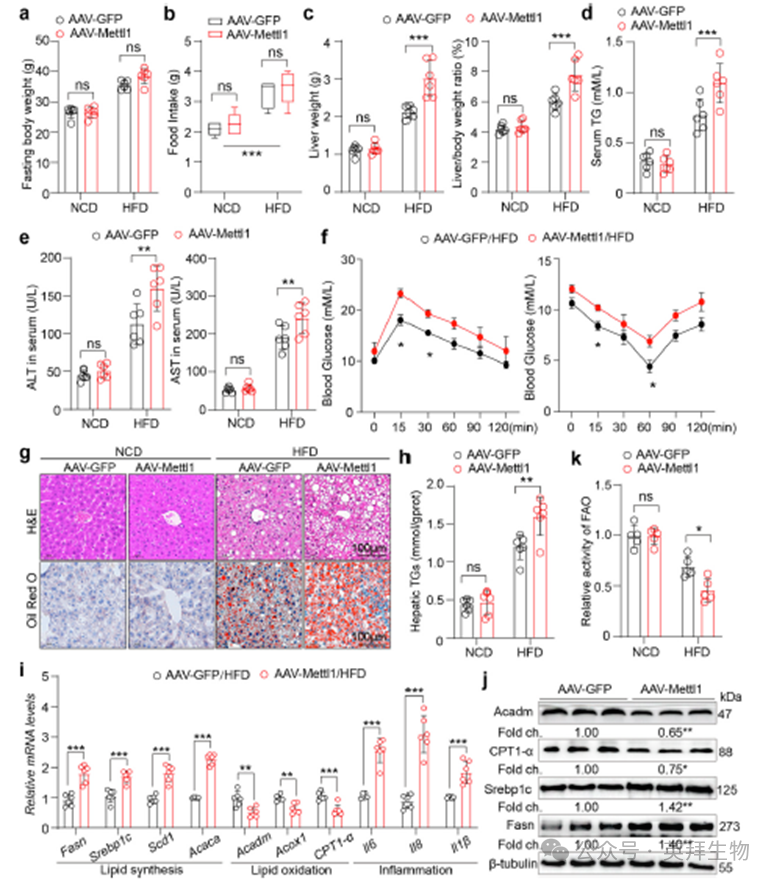
为了进一步研究Mettl1在MASLD中的作用,我们通过尾静脉注射含有Mettl1基因的腺相关病毒(AAV)构建了Mettl1过表达小鼠模型。我们观察到,无论是在NCD还是HFD条件下,Mettl1过表达小鼠与对照组在体重和食物摄入量上均无显著差异(图3a,b)。然而,在HFD组中,过表达组的肝脏重量及肝重与体重比均显著高于对照组,而在NCD组中则未观察到差异(图3c)。此外,转氨酶水平检测显示,Mettl1过表达组的ALT和AST均升高,这表明Mettl1过表达损害了MASLD小鼠的肝功能(图3e)。GTT和ITT的结果表明,Mettl1过表达导致肝脏组织的胰岛素抵抗加剧(图3f)。组织病理学评估,包括 hematoxylin-伊红(H&E)和油红O染色,以及血清和肝脏甘油三酯(TG)水平的测定,均表明在HFD喂养下,与对照组相比,Mettl1过表达促进了肝脏中的脂质积累(图3d, g,h)。进一步的qPCR分析显示,在HFD诱导的小鼠中,脂质合成相关基因的表达升高,脂肪酸氧化相关基因的表达降低,并且炎症反应增强(图3i)。蛋白质印迹分析证实,Mettl1过表达刺激了脂质合成,同时抑制了脂肪酸氧化(图3j)。此外,FAO实验证实,Mettl1过表达抑制了HFD小鼠肝脏中的脂肪酸氧化(图3k)。总之,Mettl1过表达通过促进脂质合成和抑制脂质氧化,加剧了MASLD的进展。
4)METTL1调节脂肪酸诱导肝细胞的脂肪酸合成和氧化
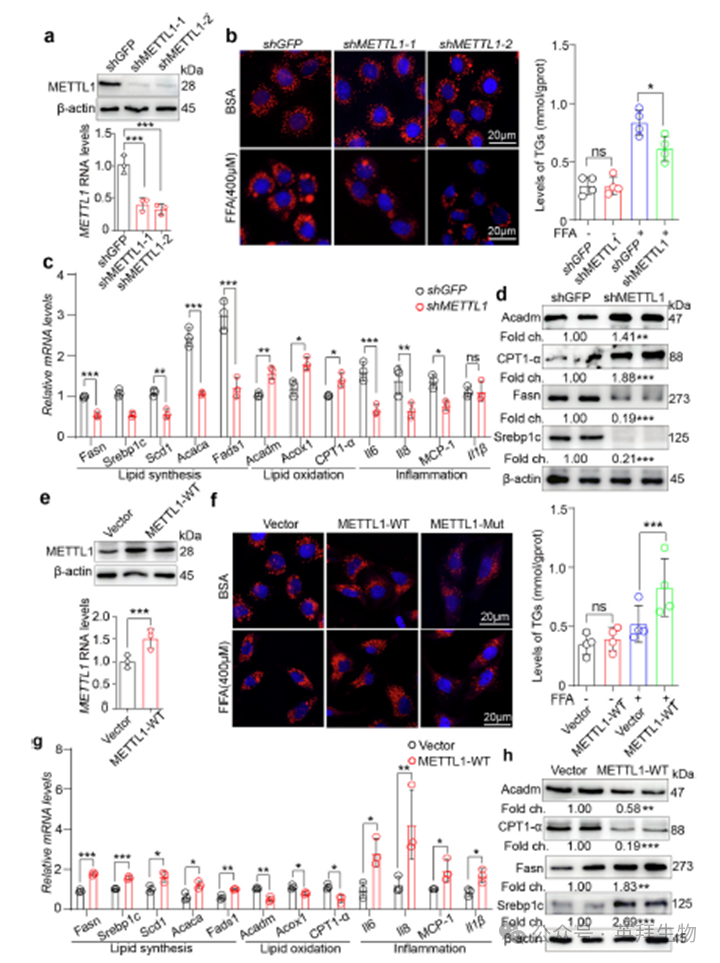
接下来,我们研究了METTL1对体外暴露于FFA的肝细胞脂质代谢和炎症的影响。我们构建慢病毒来降低METTL1的表达,并感染HepG2细胞系(图4a)。与对照组(shGFP)相比,在FFA处理下,METTL1缺失显著降低了脂质积累和TGs含量(图4b)。qPCR分析显示,METTL1的敲低抑制了与脂质合成和促炎反应相关的基因的表达,同时增强了与脂质氧化相关的基因的表达(图4c)。蛋白水平评估进一步证实,METTL1敲低抑制脂质合成并促进脂质氧化(图4d)。METTL1的过表达显著增强了FFA诱导的HepG2细胞中的脂质沉积,而突变体METTL1的过表达并未诱导这种作用(图4e,f)。分子分析显示,METTL1促进脂质合成和炎症反应,同时抑制脂质氧化(图4,h)。总的来说,METTL1在FFA诱导的肝脂肪变性中起关键作用。
5)METTL1调节FFAs诱导肝细胞内m7G修饰mRNA的转录组格局
METTL1是一种稳定的m7G甲基转移酶,mRNA中内部m7G的存在强调了这种修饰的潜在功能。为了确定METTL1是否在MASLD发病过程中调控内部m7G mRNA修饰,我们对HepG2细胞在不同条件下,包括BSA/ shGFP、FFA/shGFP和FFA/shMETTL1进行了m7G MeRIP-Seq(图5a)。我们观察到m7G峰富集在shGFP组的5 ' UTR上,位于非常靠近翻译起始位点的位置。相比之下,在经FFAs处理的shMETTL1细胞中,m7G峰不太明显,主要位于编码序列(CDS)区域(图5b)。为了进一步验证m7G在整个转录本中的分布模式,我们生成了m7G内部读取的剖面,并一致发现m7G主要位于CDS区域,以及起始密码子和5 ' UTR内(图5c)。为了探究m7G修饰与基因表达之间的关系,我们将m7G峰值数据与RNA-seq基因表达数据进行了比较。我们将上调的峰分类为高甲基化的m7G峰,其中包括高下调和高上调的基因,而下调的峰被称为低甲基化的m7G峰(图5d)。随后,通过GO分析,发现m7G修饰的mRNA富集与脂质代谢相关的功能(图5e)。此外,在比较FFA/shGFP和FFA/shMETTL1细胞时,我们发现含有m7G的差异修饰mRNA在脂质代谢功能上也富集(图5f)。有趣的是,来自三种类型细胞的内部mRNA m7G的基序分析显示出对AG或GC丰富区域的相似偏好(图5g)。此外,我们分析了BSA/shGFP和FFA/shGFP之间,以及FFA/shGFP和FFA/shMETTL1之间m7G差异mRNA位点,确定了多个与脂质代谢相关的mRNA进行了m7G修饰(图5h)。这表明METTL1介导内部mRNA的m7G修饰,从而调节脂质代谢。
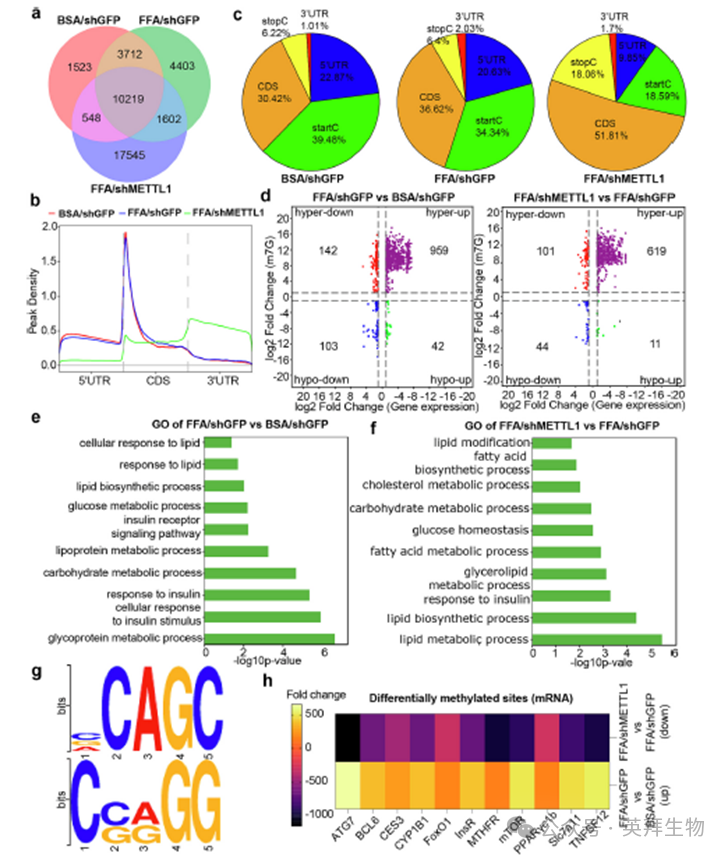
6)内部mRNA m7G促进FoxO1的稳定性和翻译
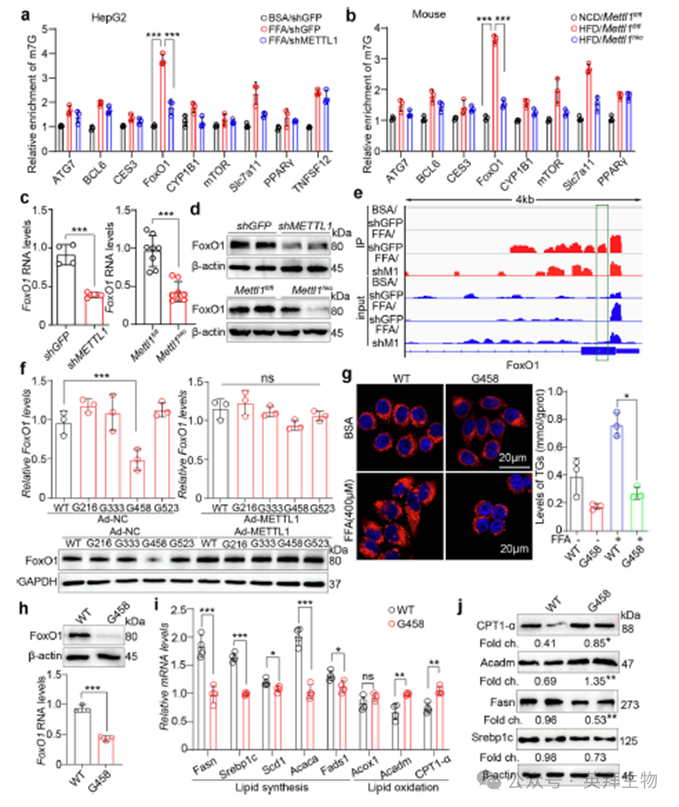
我们对具有差异甲基化位点的候选mRNA进行了MeRIP-qPCR分析,然后进行了m7G-seq分析。结果表明,在HepG2细胞中,FFA/shGFP细胞中FoxO1表达升高,而FFA/shMETTL1细胞中FoxO1表达降低(图6a)。此外,小鼠肝脏的MeRIP-qPCR分析表明,喂食HFD的小鼠FoxO1表达升高,而肝细胞特异性Mettl1敲除小鼠FoxO1表达降低(图6b)。我们随后观察到shMETTL1人肝细胞和mett1hko小鼠肝脏中FoxO1 mRNA和蛋白的表达均下降。这表明缺乏METTL1会损害FoxO1 mRNA的稳定性,导致蛋白水平降低(图6c,d)。m7G-seq分析表明FoxO1的差异m7G修饰发生在外显子1的300 bp内(图6e)。通过整合m7G位点和基序分析,我们在不改变蛋白序列的情况下,构建了人类FoxO1在G216、G333、G458和G523位点的突变质粒。研究结果显示G458突变导致FoxO1 mRNA和蛋白表达水平下降,而METTL1过表达增强FoxO1表达(图6f)。此外,我们使用过表达WT FoxO1和G458突变FoxO1的HepG2细胞进行了代谢特性分析。G458突变显著降低了FoxO1的RNA和蛋白表达水平(图6小时)。Nile Red染色和TG分析显示FoxO1 G458突变体减少了脂质积累(图6g)。此外,G458突变抑制了脂质合成相关基因的表达,同时增强了脂质氧化相关基因的表达(图6i,j)。这些数据表明FoxO1介导的脂质合成和氧化是通过METTL1介导的m7G修饰在人G458/小鼠G449外显子1位点进行调控的。
7)FoxO1过表达抵消了METTL1缺乏对MASLD脂质代谢紊乱的保护作用
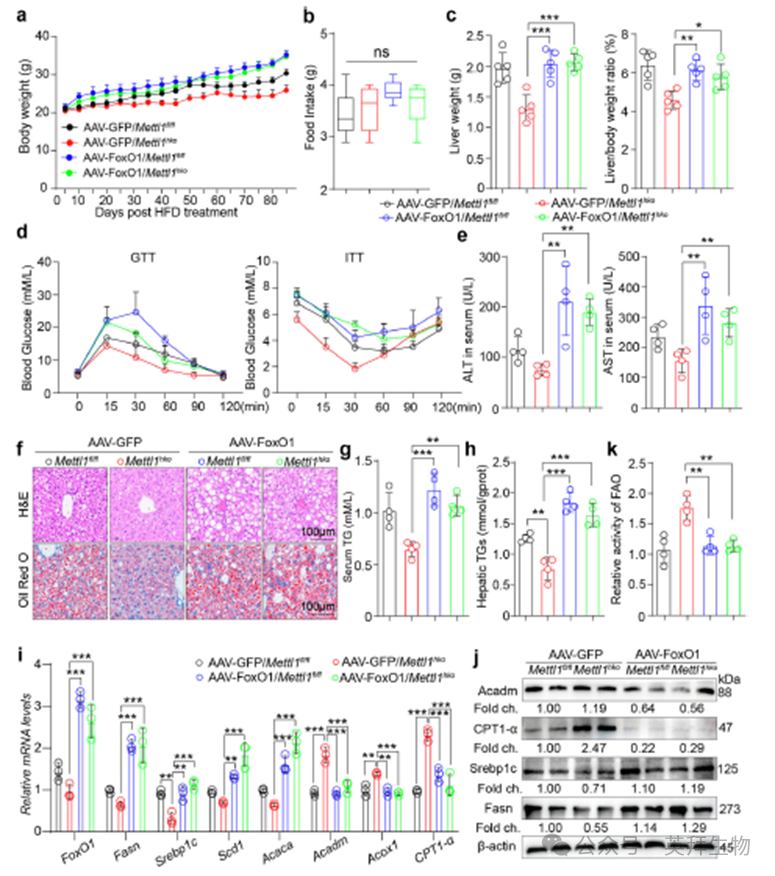
接下来,我们利用AAV在Mettl1fl/fl和Mettl1hko小鼠的肝脏中过表达FoxO1,评估其对Mettl1缺乏对MASLD的保护作用的调控作用。虽然FoxO1-过表达小鼠的摄食量与其他组没有显著差异,但在HFD喂养16周后,它们的体重大于感染GFP病毒的对照组(图7a,b)。然而,过表达FoxO1显著增加肝脏重量和肝体重比(图7c)。此外,FoxO1过表达降低了Mettl1缺乏对MASLD小鼠胰岛素抵抗的保护作用(图7d)。肝转氨酶检测显示FoxO1过表达会升高转氨酶水平,从而损害肝功能(图7e)。组织学染色和TG分析显示,Mettl1hko小鼠的肝脏脂质积累明显低于Mettl1fl/fl小鼠,而FoxO1过表达显著增加了肝脏脂质积累(图7f,g,h)。随后,我们检测了肝组织中脂质合成和氧化相关基因的表达。结果显示FoxO1过表达抵消了Mettl1缺乏对脂质代谢的影响(图7i,j)。此外,FAO实验表明FoxO1过表达可以抑制METTL1缺乏引起的脂质氧化增加(图7k)。综上所述,Mettl1缺乏对MASLD脂质代谢失调的保护作用是通过调节FoxO1的表达来介导的。
8)METTL1抑制剂对HFD诱导的MASLD有保护作用
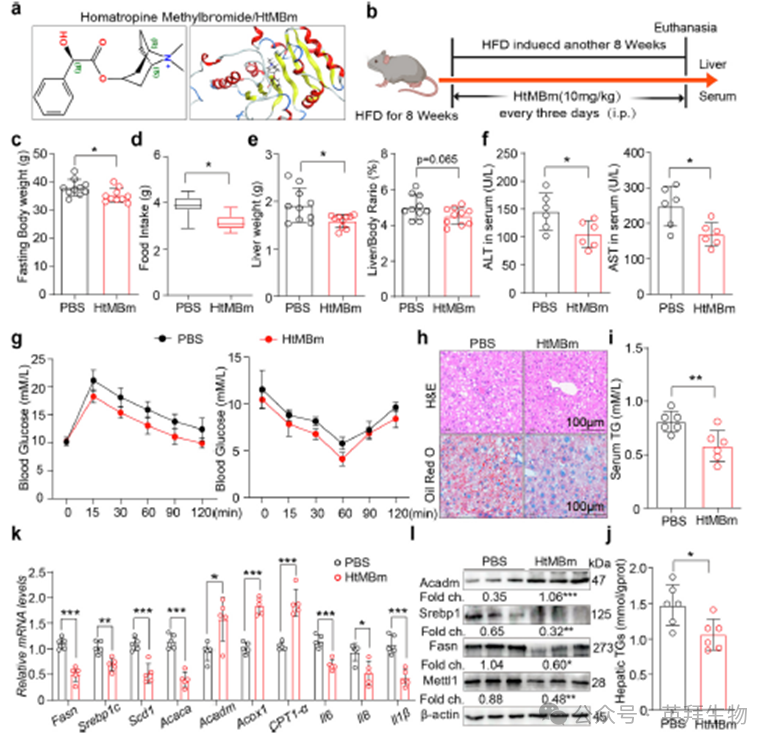
为了确定潜在的METTL1抑制剂,我们对Chemdiv2019数据库进行了基于结构的虚拟筛选,该数据库包含1,535,478种药物样化合物。从这次筛选中,我们选择了在METTL1的催化口袋中表现出最高对接分数的前10个候选化合物,最后确定HtMBm。为了进一步研究HtMBm在MASLD中的作用,我们通过HFD小鼠腹腔注射HtMBm建立了脂肪肝模型(图8b)。我们观察到,HtMBm治疗组的体重、肝脏重量、肝脏体重比和摄食量均略低于对照组(图8c,d,e)。肝转氨酶水平显示,HtMBm治疗组ALT和AST均降低,提示肝功能改善(图8f)。此外,GTT和ITT表明,HtMBm治疗可改善肝组织的胰岛素抵抗(图8g)。组织病理学评估,包括H&E和油红O染色,以及血清和肝脏TG水平的测量,表明HtMBm治疗组与对照组小鼠相比,肝脏中的脂质积累减少(图8h,i,j)。使用qPCR的进一步分析显示,脂质合成相关基因的表达减少,脂肪酸氧化相关基因的表达增加,炎症反应减少(图8k)。Western blot分析证实,HtMBm处理刺激脂质氧化,同时抑制脂质合成(图8l)。这些发现表明,HtMBm通过靶向METTL1抑制脂质合成和促进脂质氧化来改善MASLD的进展。
结论:
本研究详细了解了METTL1在MASLD发展中的功能和潜在的分子机制。这些发现为开发m7G修饰靶向治疗MASLD的临床治疗提供了有价值的见解。
参考文献:
Du J, Li Y, Zhu X, Gao J, Zhang Y, Wang C, Han D, Qiao L, Kou B, Guo R, Zhang H, Lin J. METTL1-mediated m7G methylation of FoxO1 regulates lipid metabolism in metabolic dysfunction-associated fatty liver disease. Metabolism. 2025 Oct 14;174:156420. doi: 10.1016/j.metabol.2025.156420.