






肝癌治疗新策略:靶向CAPG,通过促进铁死亡克服索拉非尼耐药
肝细胞癌(HCC)因其侵袭性肿瘤进展和有限的治疗选择,已成为全球治疗难题。因此,亟需发现新的治疗靶点。本研究通过蛋白质组测序、公共数据库分析及免疫组化技术,鉴定出CAPG作为HCC组织中上调的候选基因,且与不良临床预后相关。通过细胞系、异种移植模型及肺转移模型,我们深入探究了CAPG在HCC肿瘤发生中的生物学作用。研究发现,CAPG缺失可同步抑制体内外肿瘤增殖与转移。功能实验进一步验证了CAPG对索拉非尼诱导铁死亡的影响。通过集落形成实验、IC50测定、qPCR及Western blot分析,我们揭示了CAPG表达与索拉非尼治疗的关联性:该蛋白在药物暴露后显著上调,并参与介导耐药机制。进一步采用RNA测序、ChIP测序、免疫共沉淀及泛素化实验,阐明了CAPG的分子调控通路。机制上,CAPG通过诱导WDR74转录促进基因表达,进而调控p53与MDM2的相互作用导致p53降解。我们的研究表明,CAPG通过抑制铁死亡促进肿瘤增殖并诱导索拉非尼耐药,这表明CAPG可能是治疗HCC的潜在靶点。该研究于2025年8月发表在《International Journal of Biological Sciences》,IF:10.0。
技术路线

主要研究结果:
1.CAPG在肝细胞癌组织中上调,并与预后不良相关
为鉴定与HCC相关的差异表达蛋白(DEPs),我们对五对原发性HCC组织及其邻近正常组织进行了蛋白质组学分析。热图和火山图显示,与邻近正常组织相比,CAPG在HCC组织中属于上调幅度最大的差异表达蛋白之一(图1A、B)。完整数据集详见表S4。为验证这些发现,我们利用TCGA数据库(图1C)及三个公开的GEO数据集(GSE54236、GSE14520和GSE121248)(图S1A-C)评估了HCC和肿瘤周围组织中的CAPG表达水平。这些数据集均显示HCC组织中CAPG表达显著升高。在TCGA队列中,CAPG高表达还与总生存期缩短相关(图1D)。此外,我们通过组织微阵列分析检测了77对HCC与邻近非肿瘤组织的CAPG表达,证实其在肿瘤样本中过度表达(图1E、F)。Kaplan-Meier生存分析进一步显示,CAPG水平升高的患者总生存期(OS)和无进展生存期(PFS)显著更差(图1G、H)。
随后,我们通过qPCR和Western blotting检测了多个HCC细胞系的CAPG表达水平。结果显示97H和LM3细胞中CAPG表达相对较高,而Huh7和PLC细胞中表达较低(图S1D,E)。基于此,选取97H和LM3细胞进行稳定敲低实验,同时利用Huh7和PLC细胞建立稳定过表达细胞系以进行后续功能分析。
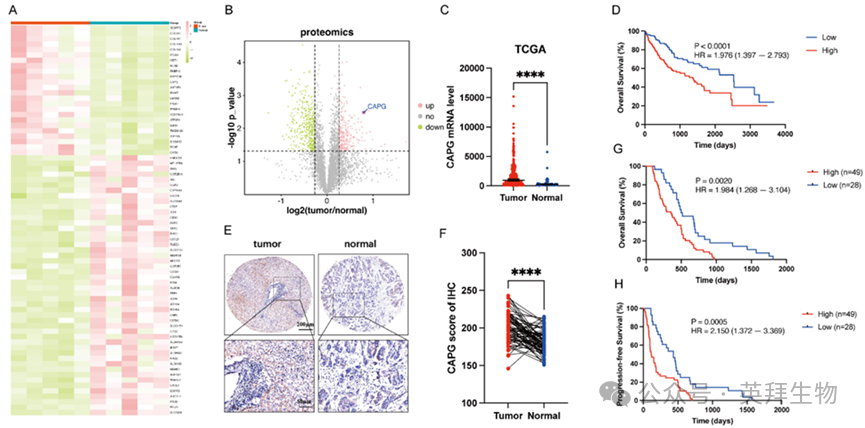
图1.CAPG在肝细胞癌组织中上调,并与预后不良相关
2.CAPG敲低抑制肝细胞癌细胞在体外和体内的增殖与转移
为探究CAPG在HCC细胞增殖、迁移和侵袭中的作用,我们首先通过检测CAPG蛋白水平验证了其敲低与过表达的有效性(图2A、S2A)。CCK-8增殖检测、集落形成实验及EdU标记实验均表明,CAPG沉默显著降低了97H和LM3细胞的存活率。相反,在Huh7和PLC细胞中过表达CAPG则提升了细胞存活率(图2B-D,S2B-D)。Transwell迁移实验、伤口愈合实验及Matrigel侵袭实验结果一致表明:CAPG敲低显著抑制97H和LM3细胞的迁移与侵袭能力,而CAPG过表达则增强这些恶性表型(图2E-G,S2E-G)。这些结果表明CAPG在体外促进HCC细胞增殖、迁移和侵袭。
为进一步评估CAPG的体内致瘤作用,将转导对照shRNA(shNC)或靶向CAPG的shRNA(shCAPG)的LM3细胞皮下注射至BALB/c裸鼠体内。CAPG敲低显著抑制肿瘤重量和体积(图3A-D)。免疫组化分析显示,源自shCAPG细胞的肿瘤中Ki67表达降低(图3E,F)。
为评估CAPG对体内转移的影响,将LM3 shNC或shCAPG细胞经静脉注射至BALB/c裸鼠体内。5周后处死小鼠并收集肺组织。组织学分析显示,与对照组相比,shCAPG组肺转移结节数量显著减少(图3L、M)。综上所述,这些发现表明CAPG在体外和体内均对促进HCC增殖和转移起关键作用。
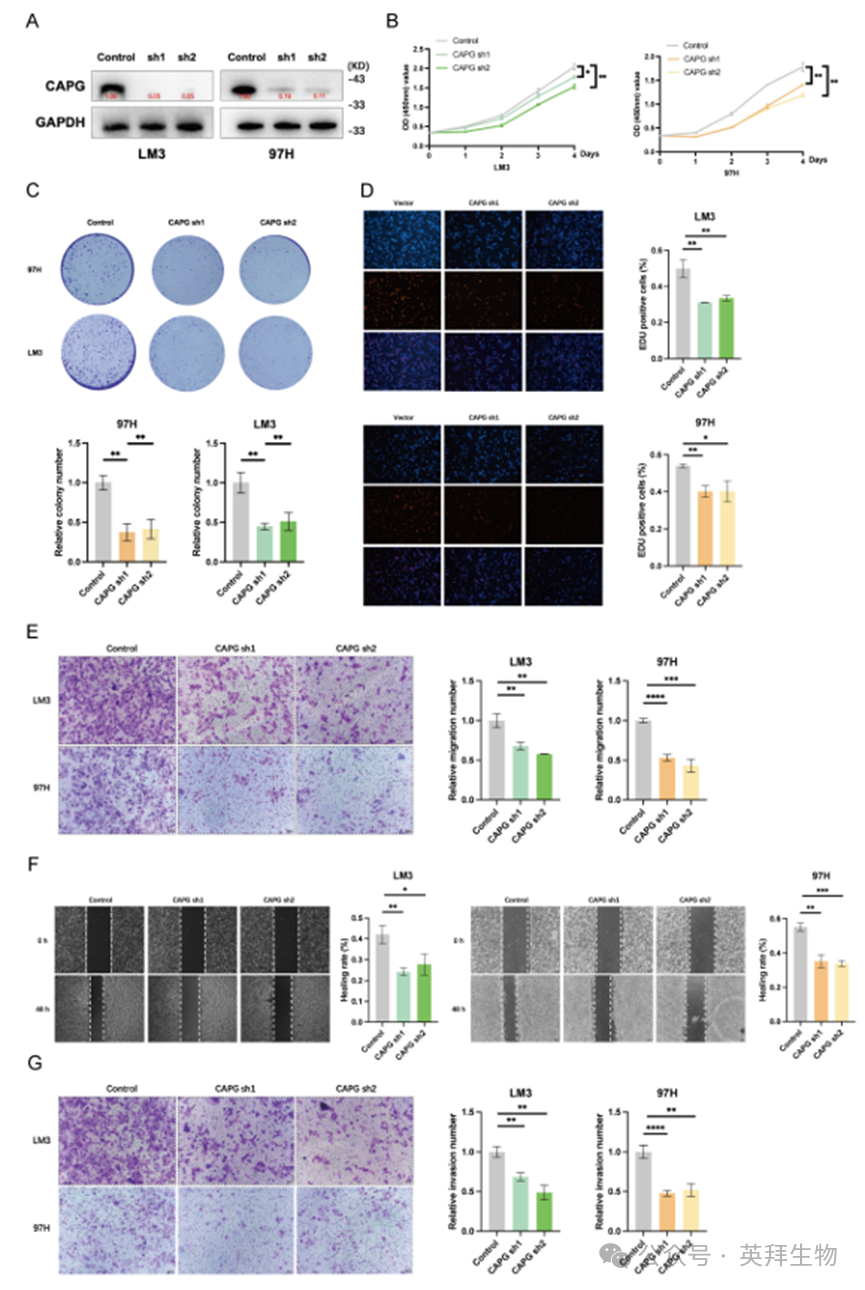
图2.CAPG促进HCC细胞增殖、迁移和侵袭
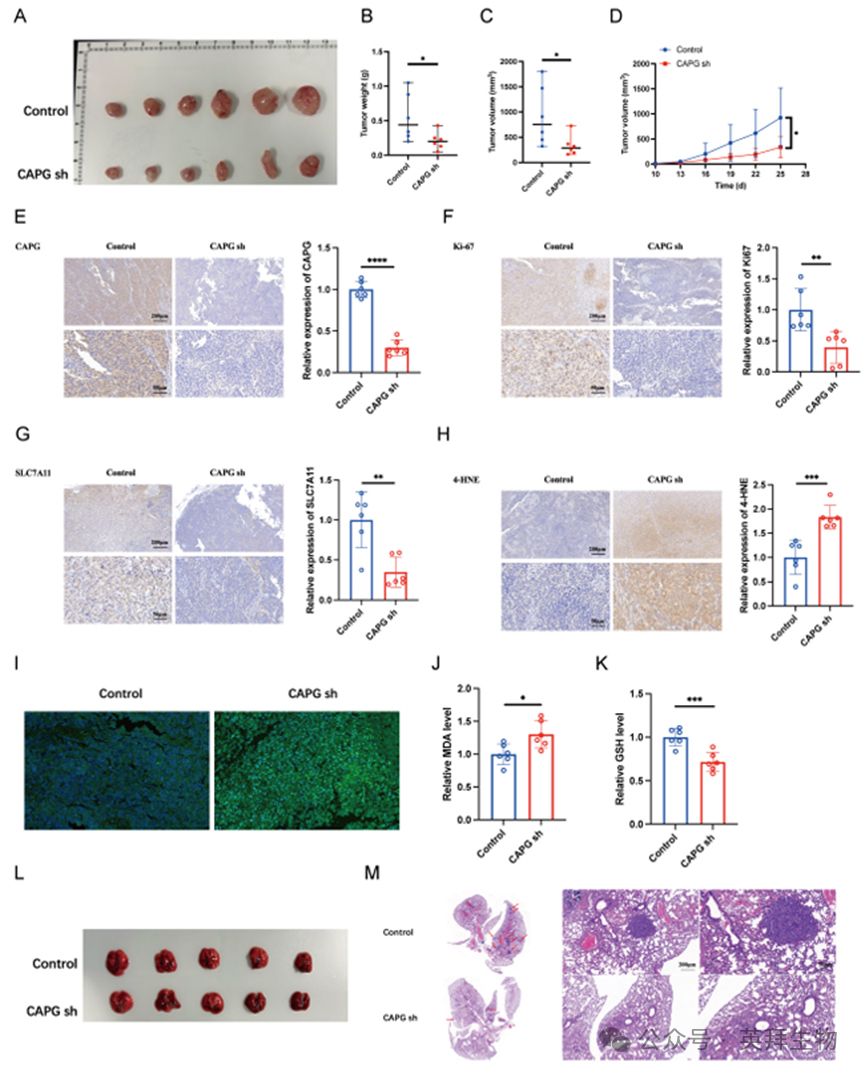
图3.CAPG沉默通过诱导体内铁死亡抑制肝细胞癌肿瘤增殖与转移
3.CAPG过表达抑制索拉非尼诱导的肝细胞癌细胞铁死亡
鉴于CAPG的mRNA和蛋白表达受到Erastin显著调控,我们探究了CAPG是否通过调控铁死亡促进肝细胞癌(HCC)进展。为评估肿瘤组织中的铁死亡水平,我们进行了SLC7A11和4-HNE的免疫组化染色,并测定了脂质过氧化、丙二醛(MDA)水平及谷胱甘肽(GSH)含量。值得注意的是,shCAPG肿瘤表现出SLC7A11和4-HNE表达降低(图3G、H),脂质过氧化(图3I)和MDA水平升高(图3J),以及GSH水平下降(图3K),表明铁死亡增强。
索拉非尼可作为HCC的铁死亡诱导剂。本研究成功建立SR 97H和Huh7细胞系,并通过IC50测定、CCK-8检测、qPCR及Western blot验证其耐药性(图S3A-D)。我们进一步证实索拉非尼可在亲本细胞系及SR耐药细胞系中诱导铁死亡,表现为MDA水平升高和脂质过氧化增强(图S4A,B)。基于这些发现,我们随后探索了CAPG表达与索拉非尼诱导铁死亡之间的潜在关联。
我们首先在HCC细胞中验证了经索拉非尼(10 μM)孵育24小时后敲低和过表达CAPG的有效性(图4A、S5A)。线粒体形态学改变(如体积缩小、嵴结构消失及膜密度增加)是铁死亡的经典特征。透射电子显微镜显示,CAPG敲低加剧了这些线粒体异常并增强了索拉非尼诱导的细胞死亡,而CAPG过表达则减轻了这些效应(图4B)。随后我们评估了其他铁死亡相关参数,包括脂质过氧化、细胞内铁含量、丙二醛水平及谷胱甘肽水平。CAPG敲低导致脂质过氧化、铁含量和丙二醛水平升高,同时谷胱甘肽水平降低。相反,CAPG过表达产生相反效应(图4C-F,S5B-E)。我们进一步检测了索拉非尼处理(10μM,24小时)后铁死亡相关基因的mRNA和蛋白表达。CAPG敲低降低了铁死亡抑制基因SLC7A11的表达,同时增加了铁死亡促进基因TFRC和ACSL4的表达。相反,CAPG过表达则逆转了这些效应(图4G、H,S5F、G)。
在使用已知铁死亡诱导剂兼SLC7A11抑制剂——10μM的Erastin处理后,观察到类似结果(图4I、S5H),该抑制剂与索拉非尼具有相同的分子靶点。随后我们探究了敲低CAPG是否能增强SR型肝癌细胞对铁死亡的敏感性。如预期所示,SR细胞对Erastin的响应弱于索拉非尼敏感细胞。然而敲低CAPG后,SR细胞对Erastin的敏感性得以恢复(图S6A,B)。此外,在耐药细胞中沉默CAPG后,经索拉非尼处理可导致脂质过氧化和丙二醛水平升高,同时谷胱甘肽水平下降(图4J-L)。
敲低CAPG还增强了SR细胞对索拉非尼的敏感性,表现为细胞活力下降。这种增敏效应可被铁死亡抑制剂Fer-1(Ferrostatin-1)联合治疗逆转(图4M)。为进一步验证铁死亡的参与作用,我们比较了多种细胞死亡抑制剂的效果。仅Fer-1能缓解索拉非尼诱导的细胞死亡,而铜死亡抑制剂TTM、凋亡抑制剂Z-VAD-FMK、坏死抑制剂坏死硫酰胺、活性氧抑制剂N-乙酰半胱氨酸及自噬抑制剂氯喹均无效(图S6C)。这些发现表明,索拉非尼处理后CAPG敲低诱导的细胞死亡增加是由铁死亡介导的。
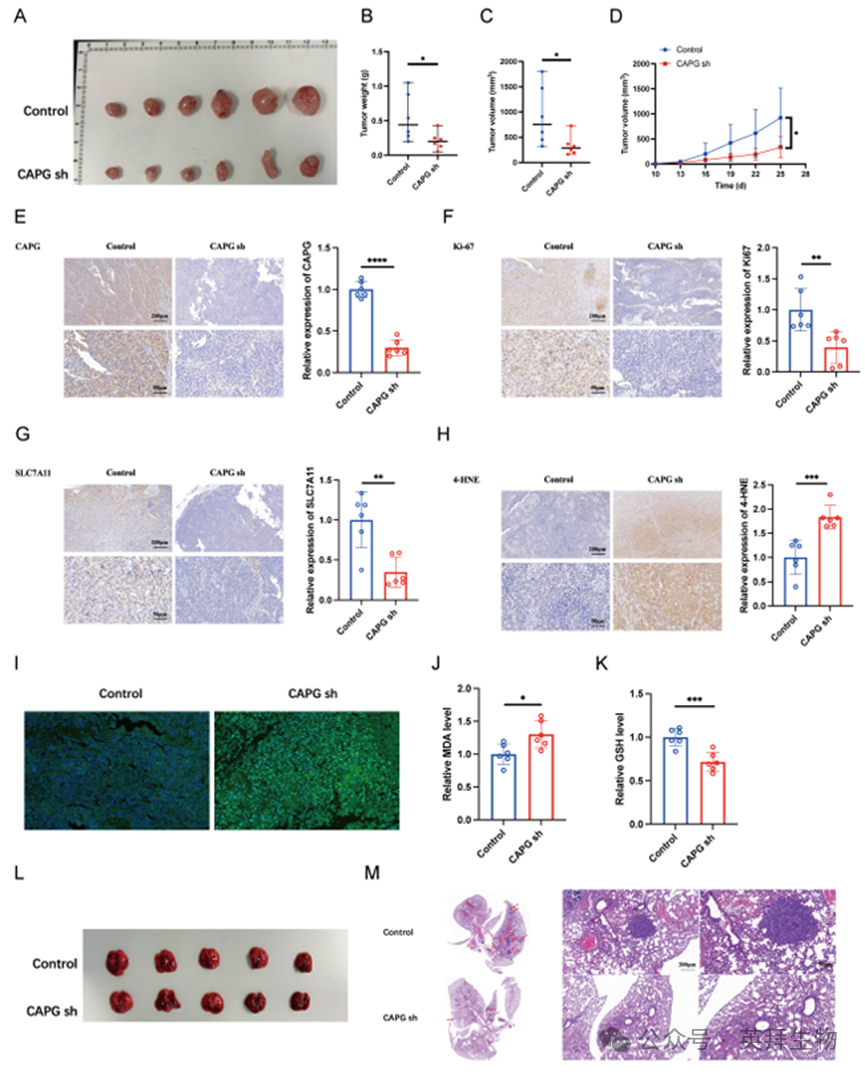
图4.CAPG通过调控索拉非尼诱导的铁死亡在肝细胞癌细胞中的作用
4.CAPG促进HCC细胞对索拉非尼的耐药性,且在索拉非尼治疗后其表达上调
接下来,我们探讨了CAPG与索拉非尼耐药性的关联。CCK-8和集落形成实验表明,CAPG敲低显著增强了索拉非尼诱导的细胞死亡,而CAPG过表达则赋予耐药性(图5A-D)。在CAPG沉默的HCC细胞中,索拉非尼的IC50值较对照组显著降低(LM3细胞:3.842 μM vs. 6.057 μM; 97H:6.323 μM vs. 8.399 μM;图5E),而在CAPG过表达细胞中IC50值升高(PLC:5.805 μM vs. 2.740 μM; Huh7:7.839 μM vs. 4.670 μM;图5F)。SR型HCC细胞系也呈现一致结果(图S7A-C)。这些发现表明CAPG缺失可增强HCC细胞对索拉非尼的敏感性。
随后我们评估索拉非尼是否调控CAPG表达。GSE62813数据集分析显示,HepG2-SR细胞中CAPG mRNA水平较亲本细胞显著升高(图6A)。该上调现象通过qPCR和Western blot在SR细胞系中得到进一步验证(图6B,C)。组织微阵列分析还揭示CAPG表达与索拉非尼敏感性呈负相关(图6D,E)。最后,我们考察了索拉非尼处理后CAPG表达的时间动态变化。经10μM索拉非尼处理的HCC细胞呈现时间依赖性CAPG表达上升,约24小时达到峰值。然而48小时后CAPG水平开始下降,这可能是由于长期药物暴露诱导了翻译抑制(图S8)。
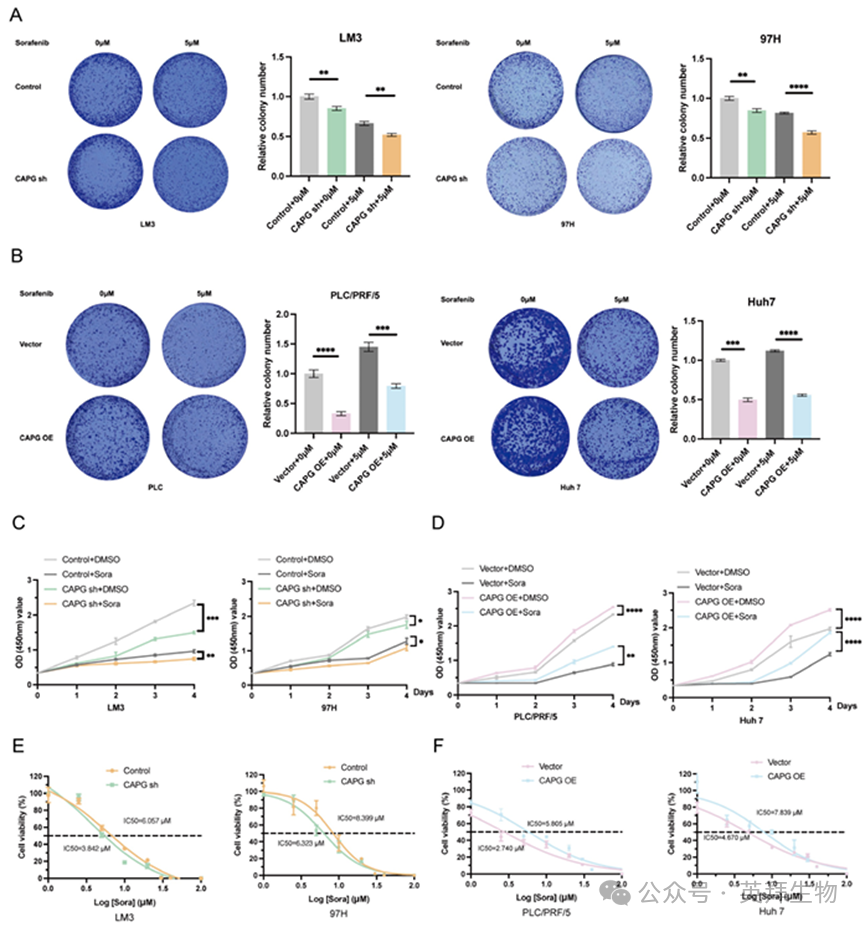
图5.CAPG促进HCC细胞对索拉非尼产生耐药性
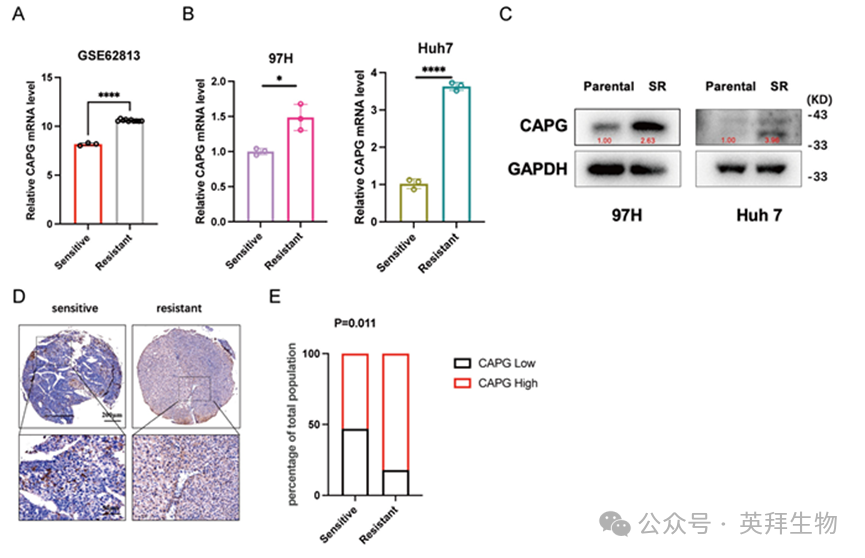
图6.索拉非尼治疗可上调HCC细胞中CAPG的表达及其向细胞核的转位
5.CAPG激活HCC细胞中WDR74的转录
为深入探究CAPG在肝细胞癌中的作用机制,我们通过RNA测序和ChIP测序技术鉴定了CAPG调控基因。RNA测序分析显示,与对照细胞相比,CAPG过表达的PLC细胞中存在2,869个上调基因和2,582个下调基因(图7A)。ChIP-seq鉴定出208个基因的峰值位于启动子转录起始位点,并确定了前五位预测的新建及已知CAPG结合基序(图7B)。通过RNA-seq与ChIP-seq数据集的交叉分析,我们锁定了54个由CAPG直接调控的候选转录靶点(图7C)。Western blotting揭示CAPG通过p53而非Nrf2或HIF-1α途径调控SLC7A11表达(图7D)。索拉非尼治疗可上调p53表达,因此我们证实WDR74与p53的相互作用并非由索拉非尼诱导的p53表达所致(图S9)。
基于上述发现,我们提出假说:先前报道在黑色素瘤中促进p53降解的WDR74基因,可能是CAPG的主要下游效应因子。TCGA数据证实了这一假说:在HCC中,CAPG与WDR74表达呈显著正相关(图7E)。ChIP-seq分析揭示CAPG在WDR74启动子区域(11号染色体62,841,380-62,841,911位点)存在结合峰(图7F)。该结合关系通过双荧光素酶报告基因实验和ChIP-qPCR进一步得到验证(图7G、H)。此外,qPCR和Western blot分析表明,敲低CAPG后WDR74表达下降,而过表达CAPG后WDR74表达上升(图7I、J)。这些结果表明,CAPG通过引导结合至WDR74启动子区域,在肝细胞癌细胞中转录激活WDR74。
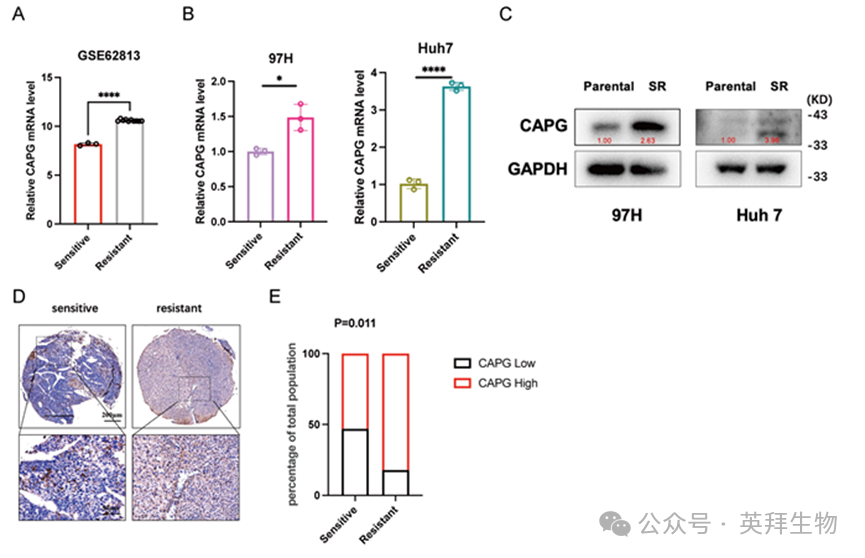
图7.CAPG参与HCC细胞中WDR74转录的激活
6.WDR74调控p53与MDM2之间的蛋白质结合
为阐明WDR74在肝细胞癌中与p53相互作用的机制,我们考察了WDR74在MDM2-p53信号轴中的作用。如图8A所示,敲低WDR74可增加p53蛋白水平并降低MDM2蛋白水平。相反,WDR74过表达则降低p53表达并提升MDM2水平。为进一步验证WDR74是否通过泛素-蛋白酶体通路调控p53降解,我们进行了共免疫沉淀(Co-IP)及p53泛素化检测。Co-IP结果显示,WDR74敲低会阻断p53与MDM2的相互作用(图8B)。与之相符的是,WDR74敲低导致p53泛素化水平降低。相反,WDR74过表达增强了p53泛素化并促进其降解(图8C)。
随后,我们探究了WDR74是否介导CAPG对铁死亡的作用。共转染WDR74质粒可部分逆转脂质过氧化、MDA水平升高及GSH水平降低的现象(图8D、F、H)。相反,敲低WDR74可减轻CAPG过表达对这些铁死亡标志物的影响(图8E、G、I)。
为深入探究CAPG-WDR74-p53-SLC7A11调控轴,我们通过慢病毒转导、siRNA转染及质粒过表达技术在肝细胞癌细胞中调控了CAPG、WDR74和p53的表达。敲低CAPG显著下调WDR74和SLC7A11蛋白水平,但WDR74过表达可逆转该效应。此外,p53过表达可抑制WDR74诱导的SLC7A11表达上调(图8J,K)。
最后,对WDR74沉默的PLC细胞进行RNA测序分析,识别出受WDR74调控的铁死亡抑制基因与驱动基因。其中SLC7A11是显著的重叠基因(图S10A)。综上所述,这些结果表明CAPG通过WDR74调控p53/SLC7A11通路,从而促进HCC细胞中铁死亡的抑制。
为进一步阐明WDR74在CAPG诱导恶性表型中的作用,进行了挽救实验。CCK-8、EdU、Transwell迁移、Matrigel侵袭实验及索拉非尼IC50分析结果表明,CAPG沉默抑制了LM3细胞的增殖、迁移和侵袭能力。然而,这些抑制效应可被WDR74过表达逆转。相反地,CAPG过表达促进了Huh7细胞的增殖、迁移和侵袭能力,而敲低WDR74则显著抑制了这些效应(图S10B-F)。
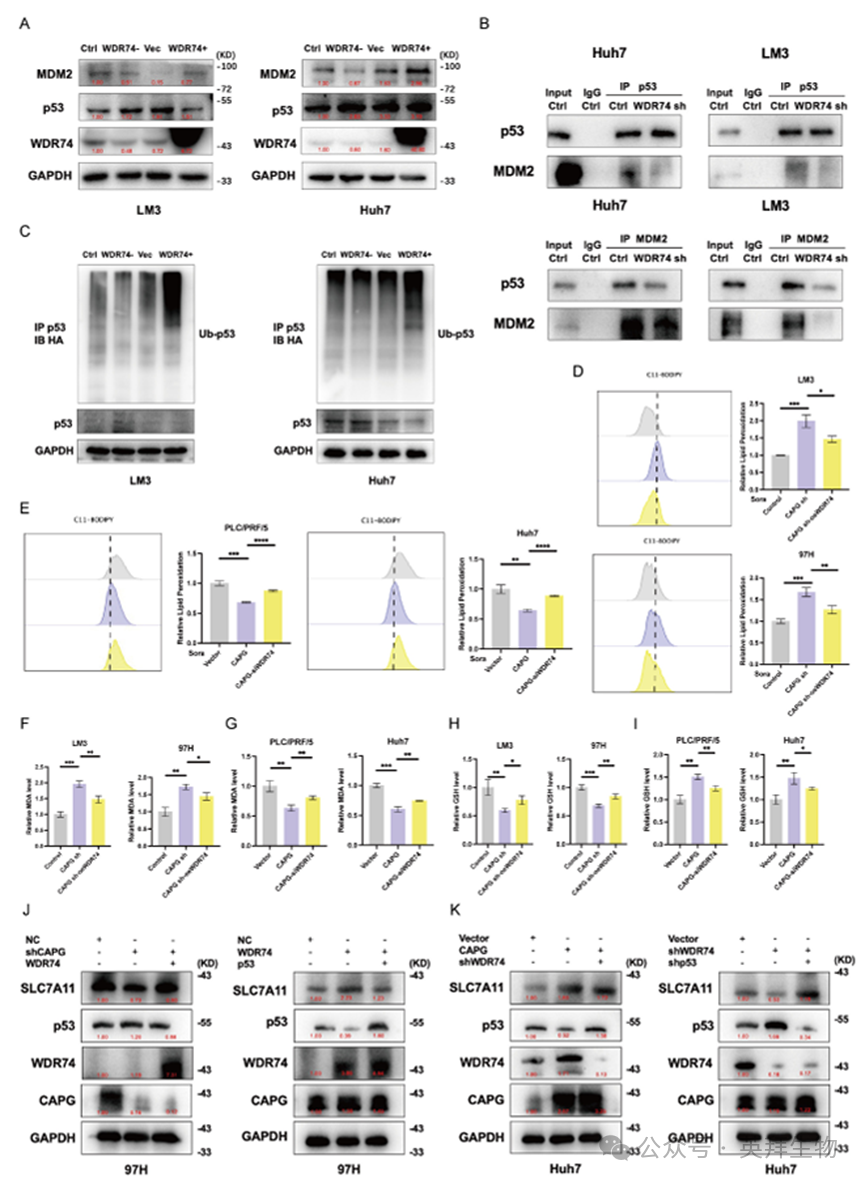
图8.敲低WDR74会破坏p53与MDM2之间的相互作用
7.CAPG/WDR74/p53/SLC7A11通路调控索拉非尼诱导的铁死亡
为验证体内实验结果,我们建立了异种移植模型,使用对索拉非尼耐药的97H细胞,通过稳定敲低CAPG、过表达WDR74、敲低SLC7A11或对照处理进行分组。索拉非尼单药治疗仅能轻度抑制体内肿瘤生长。敲低CAPG显著降低肿瘤重量、体积及生长速率。该抑制作用可被WDR74过表达部分逆转,而敲低SLC7A11则减轻了其促肿瘤效应(图9A-D)。如预期所示,铁死亡相关标志物(包括4-HNE染色、C11-Bodipy荧光强度、丙二醛水平及谷胱甘肽水平)进一步证实CAPG/WDR74/p53/SLC7A11轴参与介导索拉非尼诱导的体内铁死亡(图9E-H)。此外,我们利用CAPG过表达的Huh7细胞建立异种移植模型,进一步验证了该调控轴在体内的相关性(图S11A-I)。
为进一步验证CAPG、WDR74与p53的表达关联性,对HCC组织微阵列进行免疫组化染色。代表性图像见图9I。相关性分析表明:CAPG表达与WDR74呈正相关,与p53呈负相关;WDR74表达与p53水平亦呈负相关(图9J-L)。
尽管近年来肝细胞癌(HCC)患者的发病率和死亡率有所下降,但现有治疗策略的疗效仍不尽如人意。因此,亟需深入探索新型治疗靶点和生物标志物,以改善HCC患者的临床预后。本研究证实,CAPG通过抑制铁死亡促进HCC肿瘤增殖并增强其对索拉非尼的耐药性。机制上,WDR74被鉴定为CAPG的直接转录靶点。WDR74破坏p53与MDM2的相互作用,导致p53降解增强及SLC7A11表达上调,从而抑制铁死亡(图9M)。
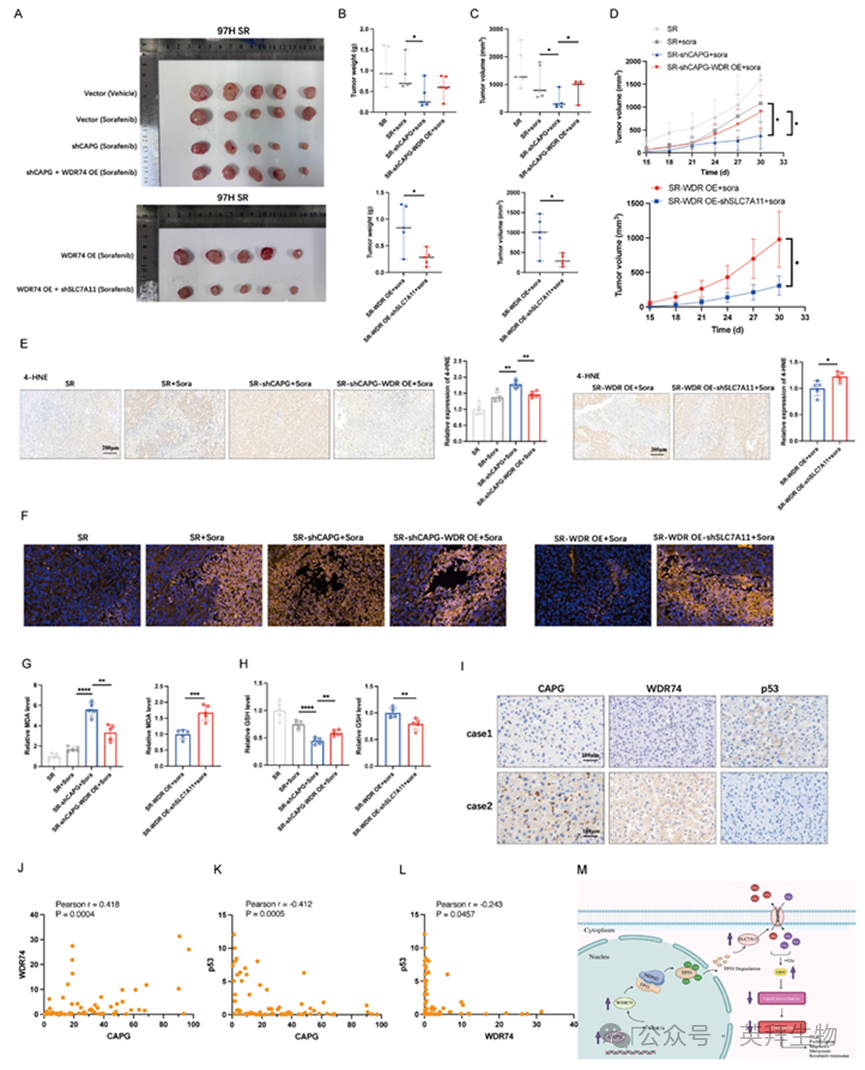
图9.CAPG/WDR74/p53/SLC7A11通路调控索拉非尼诱导的体内铁死亡
结论
综上所述,我们阐明了CAPG促进HCC肿瘤增殖和索拉非尼耐药性的特定分子机制。通过体内和体外模型研究,我们证实CAPG/WDR74/p53/SLC7A11通路通过抑制铁死亡促进HCC进展。这些发现为CAPG的致癌作用提供了新见解,并突显其作为HCC有效治疗靶点的潜力。
参考文献
Quan B, Yao F, Liu W, Tang B, Li M, Lu S, Li J, Chen R, Ren Z, Yin X. Increased CAPG inhibits ferroptosis to drive tumor proliferation and sorafenib resistance in hepatocellular carcinoma via the WDR74-p53-SLC7A11 pathway. Int J Biol Sci. 2025 Aug 22;21(12):5476-5495. doi: 10.7150/ijbs.111419. PMID: 40959275; PMCID: PMC12435477.