






新的铁死亡调控回路——蛋白激酶ATM通过磷酸化协调铁自噬

铁死亡是一种由细胞内生物活性铁催化的脂质过氧化物的致死积累驱动的程序性细胞死亡。Ser/Thr蛋白激酶ATM,DNA双链断裂损伤的主要传感器,是铁死亡执行不可或缺的。本研究中,作者发现ATM激酶支配细胞内不稳定的铁池,或者通过磷酸化铁自噬受体NCOA4,从而操纵铁自噬的铁周转。研究结果揭示了一种新的调控回路,包括ATM-NCOA 4在协调铁自噬和铁生物利用度。该研究于2023年6月发表于《Autophagy》上,IF=13.3。
技术路线









主要研究内容
1.ATM激酶在细胞铁死亡中的作用
为了证实ATM激酶的铁结合调节能力,使用ATM敲除MEF细胞。我们发现ATM敲除保护MEF免于由erastin和RSL 3触发的细胞死亡,如细胞活力测定所证明的(图1A和1B)。补充erastin或RSL 3导致WT细胞中明显的PI阳性染色,其几乎可以被Fer完全阻断。ATM敲除显著拮抗这种PI阳性染色,如荧光显微镜扫描和流式细胞术分析所示(图1C-1F)。此外,erastin和RSL 3的暴露诱导脂质过氧化物的过度产生,其在ATM敲除MEF细胞中被显著抑制(图1G和1H)。总的来说,这些数据证实了ATM激酶在铁蛋白减少过程中不可或缺的作用。此外,我们还证实了ATM激酶在其他诱导剂诱导的铁死亡中的相关性。丁硫氨酸亚砜亚胺(BSO)和柳氮磺胺吡啶(Sul)分别通过不可逆地抑制γ-谷氨酰半胱氨酸合酶(γ-glutamylcysteine synthase,γ-GS)和强有力地靶向系统Xc-,消耗细胞内GSH,从而启动铁代谢。ATM缺失也抵消了BSO和硫诱导的铁死亡,如细胞活力测定(图1I和1J)、PI染色(图1K-1N)和脂质过氧化分析(图1O和1P)所示。总之,这些发现表明ATM激酶对于铁磷酸化执行是不可或缺的。

图1:ATM激酶在细胞铁死亡中是不可或缺的
2.ATM决定的铁死亡不依赖于TRP53
肿瘤抑制因子TRP53是ATM激酶最重要的下游靶点之一。更重要的是,已报道TRP 53通过多种机制操纵铁凋亡,而不仅仅是其对细胞凋亡和自噬的充分表征的调节作用。为了研究TRP53在ATM介导的铁凋亡中的潜在作用,分析了在TRP53消融的遗传背景下ATM敲除MEFs和对照MEFs的铁凋亡敏感性。在本文中,AP29细胞是TRP53单敲除的,而AP26细胞是ATM TRP53双敲除的(图2A)。与WT MEF相比,TRP53单敲除AP29细胞在一定程度上对erastin和RSL 3诱导的细胞死亡更具抗性(图2B和2C),表明TRP53的促铁蛋白激酶活性。当与TRP53单敲除AP29细胞相比时,ATM TRP53双敲除AP26细胞似乎对erastin和RSL3诱导的铁死亡更加不敏感,如细胞活力测定(图2B和2C)、PI染色(图2D)和脂质过氧化分析(图2 E和2F)所示。值得注意的是,在暴露于erastin和RSL3的AP26细胞中观察到Ptgs2的表达降低(图2G和2 H)。为了进一步证实TRP53在ATM介导的铁磷贮积症中的相关性,使用了匹氟菊酯-α(PFTα),一种抑制TRP 53响应基因的TRP53依赖性反式激活的特异性TRP 53抑制剂。补充PFTα可明显下调Cdkn1a/p21(细胞周期蛋白依赖性激酶抑制剂1A(P21);一种重要的TRP53靶向基因)和MEF细胞中Slc7a11的上调(图2 I),表明PFTα确实抑制TRP53的转录因子活性。在PFTα攻击期间,在ATM KO细胞中观察到Cdknla的边际下调和Slc7al1的上调(图2 I),这可能是由于ATM KO细胞中TRP53的较低表达(图2A)。值得注意的是,PFTα补充在WT和ATM KO细胞中均驱动铁凋亡抗性(图2 J、2K),进一步表明TRP53的铁凋亡促进能力。另外,在WT和ATM KO细胞中,更长时间的erastin或RSL 3处理导致更多的细胞死亡。具体地,当与PFTα处理的WT细胞相比时,PFTα处理的ATM KO细胞对erastin和RSL3表现出高得多的不敏感性(图2 J、2K)。总之,这些研究表明ATM决定的细胞铁死亡,至少部分独立于TRP53。
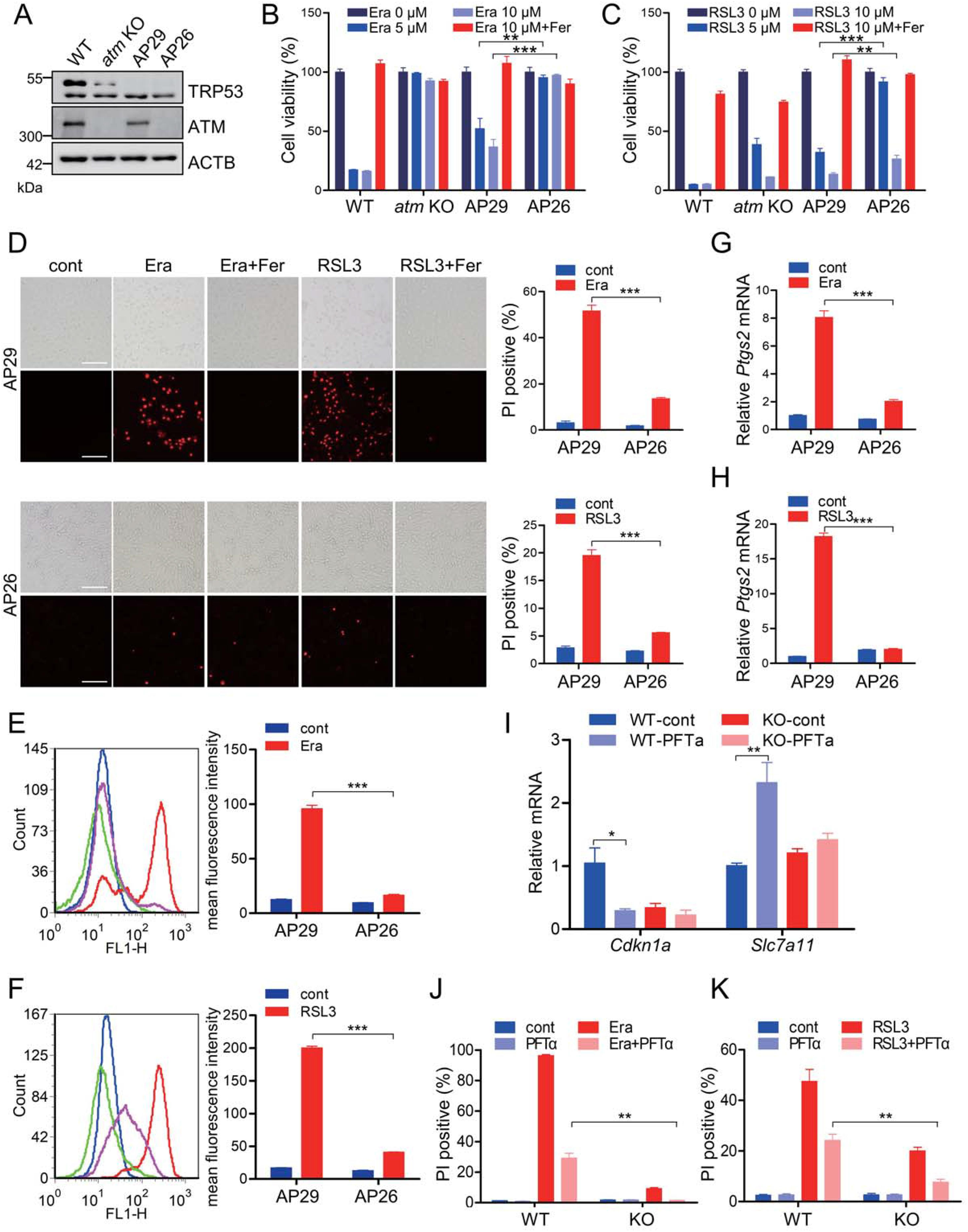
图2:ATM决定的铁死亡不依赖于TRP53
3.ATM决定了与铁蛋白相关的铁自噬
细胞内不稳定的游离铁是决定氧化还原稳态的重要因素之一。细胞可渗透的FerroOrange是一种新型的荧光铁传感器,其能够实现细胞内亚铁的活细胞荧光成像。铁凋亡诱导剂导致WT MEF细胞中FerroOrange的荧光增强,而在相同处理下在ATM KO MEF细胞中该荧光大大减弱(图3A-3C)。此外,在具有稳定ATM敲低的HT-1080中也观察到类似的表型(图3D-3F),表明ATM在铁死亡执行期间支配细胞内不稳定游离铁的观点。然而,铁外排输出蛋白SLC40A1/FPN(溶质载体家族40成员1)和铁储存相细胞溶质铁主要被铁蛋白纳米笼隔离。据报道,NCOA4介导的铁自噬在细胞铁死亡诱导期间被激活,而一般自噬组分或特异性铁自噬受体NCOA4的耗竭急剧降低铁动员,导致细胞不稳定铁库减少并拮抗铁细胞凋亡。erastin暴露导致WT MEF细胞中FTHl蛋白增加(图3G)。在WT MEF细胞中,在erastin暴露期间还观察到mRNA水平上的升高的FTHl表达,如定量RTPCR测定所示(图3I)。FTH1在ATM KO MEF细胞稳定状态下高表达。然而,在erastin暴露期间,在ATM KO细胞中未观察到FTHl蛋白或mRNA的明显增加,表明在ATM消融期间铁蛋白周转减慢(图3G和3I)。另外,这在具有RSL3处理的ATM KO细胞中是类似的(图3 H)。为了进一步证实ATM在确定铁凋亡相关的铁蛋白周转中的重要作用,利用ATM敲低HT-1080细胞,并且我们发现当ATM沉默时,FTHl也高度表达(图3 J和3 K)。类似地,erastin和RSL3暴露导致对照细胞中FTHl的增加。ATM敲低导致在erastin和RSL3暴露下HT-1080细胞中的惰性FTHl升高(图3 J和3 K)。此外,erastin暴露导致FTHl转录的明显增加,这在ATM敲低HT-1080中未观察到(图3L)。免疫荧光染色显示,erastin处理促进铁蛋白和溶酶体的共定位,而这种共定位在ATM KO MEF细胞和ATM敲低HT-1080细胞中受到明显抑制(图3M-3 P)。总之,这些数据表明,ATM在铁蛋白周转中主导了与铁死亡相关的铁自噬。
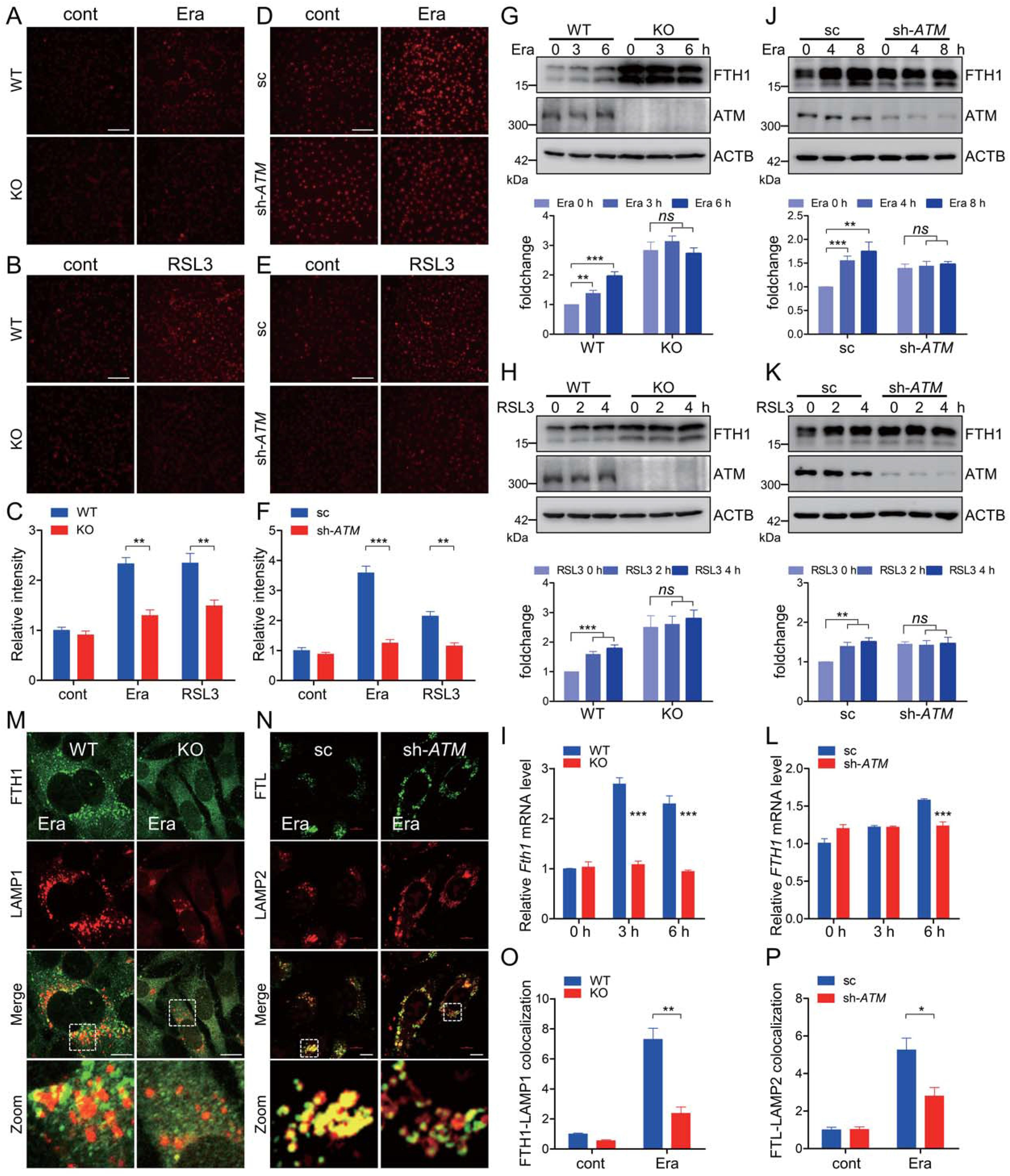
图3:ATM决定了与铁蛋白相关的铁自噬
4.ATM主导铁饥饿诱导的铁自噬
ATM是否仅操纵特异性铁蛋白沉积相关的铁自噬或ATM是否能在遗传上调节铁饥饿诱导的铁自噬是下一个重要的问题。暴露的铁螯合剂,包括去铁酮(DFP)和去铁胺(DFO),导致FTH1在MEF细胞中的快速降解。ATM敲除减弱了DFP和DFO诱导的FTH1降解(图4A和4B)。另外,ATM抑制剂KU 55933的预处理也阻断了这种FTHl降解(图4C和4D)。此外,铁螯合剂促进FTH1和溶酶体标志物LAMP1的共定位,以及铁自噬受体NCOA4和自体吞噬体标志物GFP-MAP1LC3的共定ATM的异位表达加速了DFO诱导的FTHl降解(图4G)。自噬抑制剂氯喹(CQ)急剧地阻止了这种DFO诱导的FTHl降解,即使在ATM过表达的细胞中也是如此(图4G)。研究发现,ATG9敲除和通过其抑制剂PIK-III 的PIK3C3/VPS34抑制可以在对照细胞和ATM过度表达细胞中干扰铁螯合剂诱导的FTHl降解(图4 H-4J)。这些数据进一步证实了ATM激酶在铁饥饿诱导的铁自噬中的调节作用。ATM操纵铁饥饿诱导的铁蛋白吞噬似乎是TRP53非依赖性的,因为当与TRP53单敲除AP29细胞相比时,在ATM TRP53双敲除AP26细胞中仍然观察到减慢的FTHl降解(图4K和4L)。总之,这些发现表明ATM是铁自噬的主要调节剂。然而,在ATM抑制期间,这两种共定位均被显著抑制(图4 E和4F)。总之,这些发现表明ATM是铁自噬的主要调节因子。
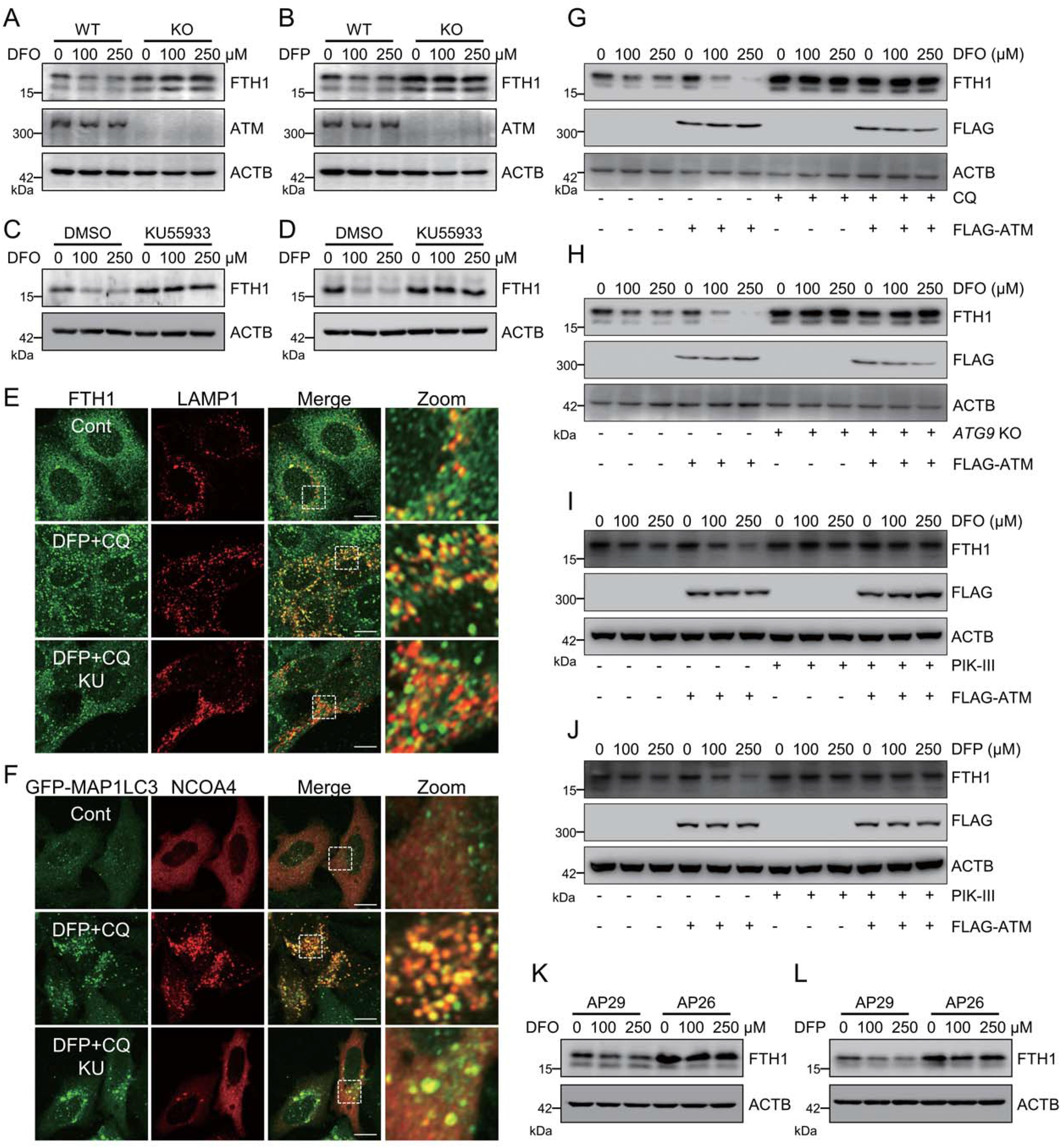
图4:ATM主导铁饥饿诱导的铁自噬
5.ATM磷酸化铁自噬受体NCOA4
稳定的NCOA4通过FTH1的保守精氨酸残基(Arg23)和NCOA4的C-末端结构域之间的直接相互作用与铁蛋白结合。在erastin补充期间,观察到NCOA 4-FTH1相互作用的明显增加,如通过免疫共沉淀分析所证明的(图5A)。在平均孵育时间,通过使用泛磷酸化丝氨酸(p-Ser)抗体检测到明显的NCOA4磷酸化。重要的是,这种NCOA4磷酸化在erastin处理期间升高(图5A)。因此,提出假设ATM激酶可能是负责这种NCOA4磷酸化。Erastin处理导致NCOA4磷酸化的升高,而当与相应的对照细胞相比时,这种磷酸化在ATM敲除MEF细胞、ATM TRP53双敲除AP26细胞和ATM敲低HT-1080细胞中几乎完全被阻断(图5B-5D),表明ATM激酶对于响应于Erastin的这种NCOA 4磷酸化是至关重要的。免疫共沉淀分析显示S550中的突变几乎消除了NCOA4磷酸化(图5E)。为了进一步证实进化上保守的S550残基(图5F)是被ATM激酶磷酸化的关键位点,通过用合成的NCOA4磷酸肽免疫小鼠,然后亲和纯化,产生针对磷酸化的S550的多克隆抗体。斑点印迹测定显示,该抗体对磷酸肽(P-肽)的特异性比对照非磷酸肽(C-肽)高得多(图5G)。通过使用该抗体,发现在对照HT-1080细胞中,响应于erastin处理,S550处的NCOA4磷酸化升高。在稳态和erastin处理状态下,在ATM沉默的HT-1080细胞中观察到S550处NCOA4磷酸化的显著降低(图5H)。总之,这些数据表明ATM在S550残基处磷酸化NCOA4。
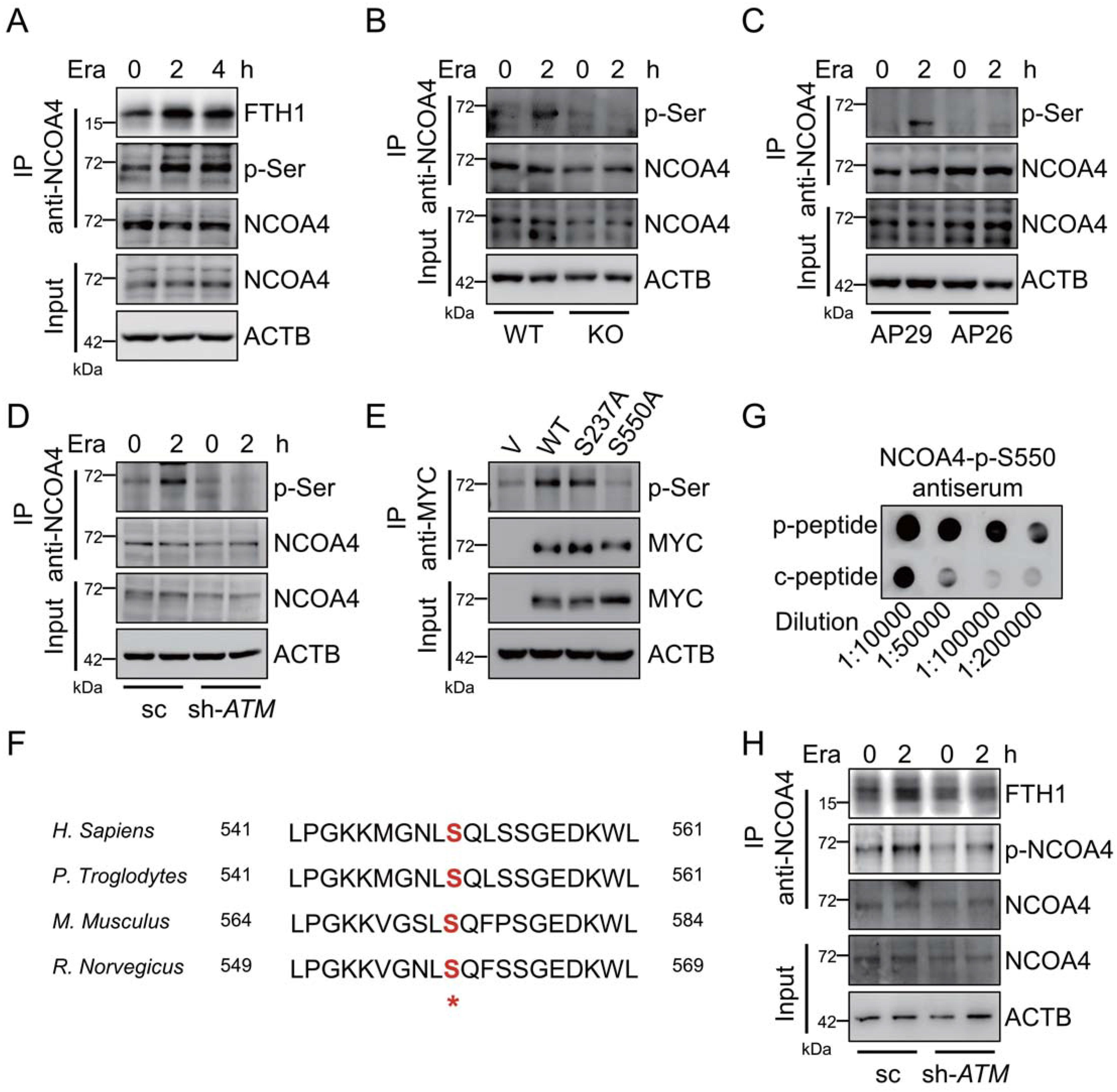
图5:ATM磷酸化铁自噬受体NCOA4
6.NCOA4磷酸化对铁自噬和铁死亡至关重要
ATM激酶介导的NCOA 4在S550残基的磷酸化是否影响铁自噬是下一个重要的问题。免疫共沉淀分析显示,S550残基中的突变明显破坏了NCOA 4-FTH1相互作用(图6A),表明ATM激酶在S550处的NCOAO4磷酸化可以增强NCOAO4与铁蛋白的相互作用。为了进一步阐明NCOAO4磷酸化在铁自噬中的相关性,构建了NCOA4稳定敲低HT-1080(图6 B)。与先前的研究一致,NCOA4沉默由于铁自噬受损而升高FTHl水平(图6 B)。此外,NCOA 4敲低深刻地消除了erastin诱导的FTHl增加,而该FTHl增加通过WT NCOA4的异位表达和S237A突变体而不是S550A突变体恢复(图6 B)。铁自噬可以更新铁蛋白,补充细胞内不稳定铁池。采用FerroOrange染色法检测细胞内不稳定铁的含量。NCOA4基因敲低后,Erastin诱导的荧光增强效应减弱。一致地,WT NCOA4的异位表达和S237A突变体而不是S550A突变体,可以恢复NCOA4敲低的HT-1080细胞中的FerroOrange荧光增强(图6C),表明在S550残基处的NCOA4磷酸化对于与铁死亡相关的铁自噬和细胞内生物活性铁的升高是至关重要的。此外,在NCOA4敲低的细胞中,erastin诱导的铁死亡被急剧消除,而WT NCOA4的异位表达和S237A突变体,而不是S550A突变体,可以恢复NCOA4敲低的HT-1080细胞中的细胞铁死亡(图6D和6 E)。类似地,WT NCOA4的异位表达而不是S550A突变体,可以恢复erastin诱导的脂质过氧化反应,这显著消除NCOA4敲低(图6 F)。总之,这些数据表明,ATM激酶在S550残基磷酸化NCOA4对于NCOA4铁蛋白相互作用、铁自噬介导的细胞游离铁升高和随后的铁死亡执行是至关重要的。

图6:NCOA4磷酸化对于铁自噬和铁死亡至关重要
结论
综上所述,这项研究表明了ATM激酶,DNA损伤反应的主传感器,是铁死亡的关键上游调节因子。在机制上,ATM激酶磷酸化铁自噬受体NCOA4,促进NCOA4-FTH1相互作用以驱动铁自噬介导的铁动员并加剧脂质过氧化。深入了解ATM激酶介导的铁蛋白代谢的病理功能,有助于开发新的治疗策略。
实验方法
细胞转染和稳转株的构建,CCK8实验,流式细胞术检测凋亡,脂质过氧化物测量,GSH测定,qRT-PCR,western blot,免疫荧光实验,铁含量测定,核质分离实验,免疫沉淀实验,克隆实验
参考文献
Wu, H., Liu, Q., Shan, X., Gao, W., & Chen, Q. (2023). ATM orchestrates ferritinophagy and ferroptosis by phosphorylating NCOA4. Autophagy, 19(7), 2062–2077. https://doi.org/10.1080/15548627.2023.2170960