






OTUD4去泛素化稳定EGFR并激活PI3K/AKT通路促进三阴性乳腺癌的侵袭
去泛素化酶 OTUD4 在多种癌症中发挥癌基因的作用,但其在三阴性乳腺癌(TNBC)中的作用尚不清楚。通过生物信息学分析和实验验证,我们证明 OTUD4 在 TNBC 中过表达,并与不良预后相关。OTUD4 表达下调可降低 TNBC 的侵袭性,突显其致癌作用。从机制上讲,OTUD4 通过稳定 EGFR 表达和激活 PI3K/AKT 通路促进 TNBC 进展。这种稳定作用通过两种机制实现:OTUD4(568–1114aa)与 EGFR(958-1210aa)之间的直接相互作用以及 OTUD4 介导的 K48 连接多聚泛素链的切割。此外,OTUD4被NRP1募集去泛素化并进一步稳定EGFR。这些发现加深了我们对 TNBC 中 EGFR 信号传导的理解,并可能为新的治疗策略提供信息。本文于2026年2月发表于Cell Death and Disease(IF=9.6)上。
技术路线:

结果:
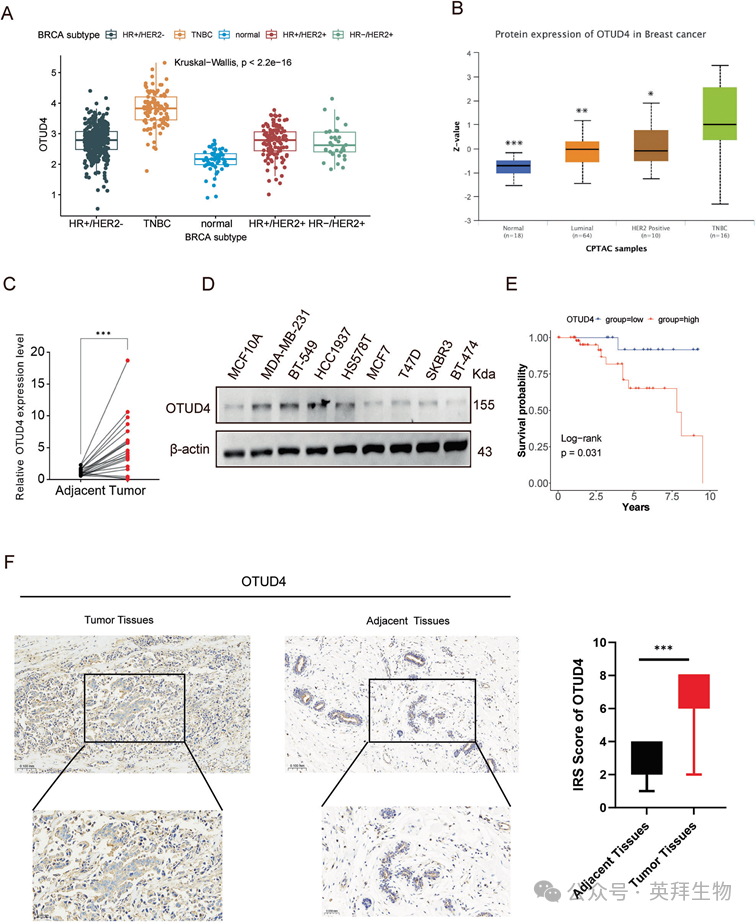
1)TNBC中OTUD4的高表达与不良预后相关
为了评估OTUD4在乳腺癌中的表达,我们分析了来自TCGA-BRCA和UALCAN数据库的数据。与正常乳腺组织相比,三阴性乳腺癌、HER2阳性乳腺癌、管腔A和管腔B亚型中OTUD4 mRNA和蛋白水平升高(图1A,B)。TNBC肿瘤组织与邻近非肿瘤组织的对比分析显示,OTUD4在TNBC中的表达明显升高(图1C)。同样,与正常乳腺上皮细胞系MCF10A和非TNBC细胞系MCF7、T47D、SKBR3、BT474相比,TNBC细胞系(MDA-MB-231、BT-549、HCC1937和HS578T)表现出更高的OTUD4蛋白水平(图1D)。TCGA数据库分析进一步表明,OTUD4表达升高与TNBC预后不良相关(图1E)。10个配对临床样本的免疫组织化学分析证实,与邻近组织相比,TNBC中OTUD4的表达更高(图1F)。基于上述结果,OTUD4可能与TNBC的预后有关。
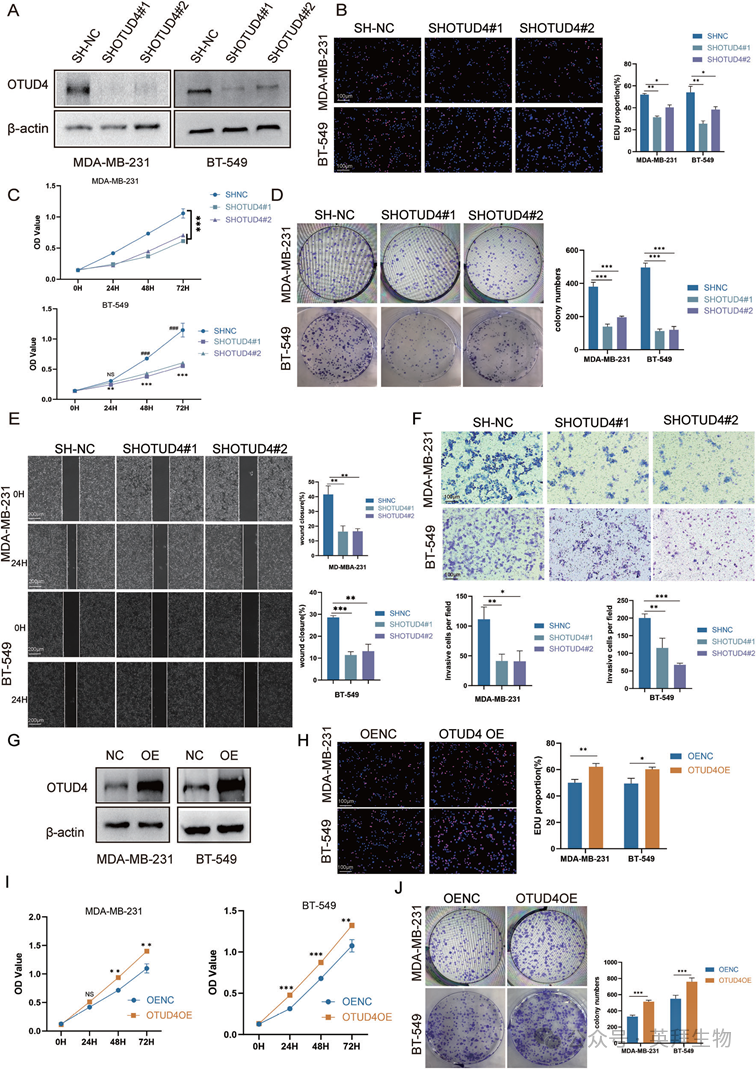
2)OTUD4在体外促进TNBC细胞的侵袭性
为了研究OTUD4在TNBC中的作用,我们在两种TNBC模型中产生了稳定的OTUD4敲低细胞系(图2A)。EDU、CCK-8和集落形成实验表明,OTUD4敲除显著降低TNBC细胞增殖(图2B-D)。在伤口愈合试验中,OTUD4敲低会损害伤口闭合,并在Transwell试验中减少TNBC细胞迁移(图2E,F)。为了进一步验证其致癌功能,我们在相同的TNBC模型中建立了过表达OTUD4的细胞系(图2G)。OTUD4过表达增强TNBC细胞增殖、集落形成(图2H-J)和迁移(图2H-J)。这些结果表明OTUD4增强了TNBC的侵袭性。
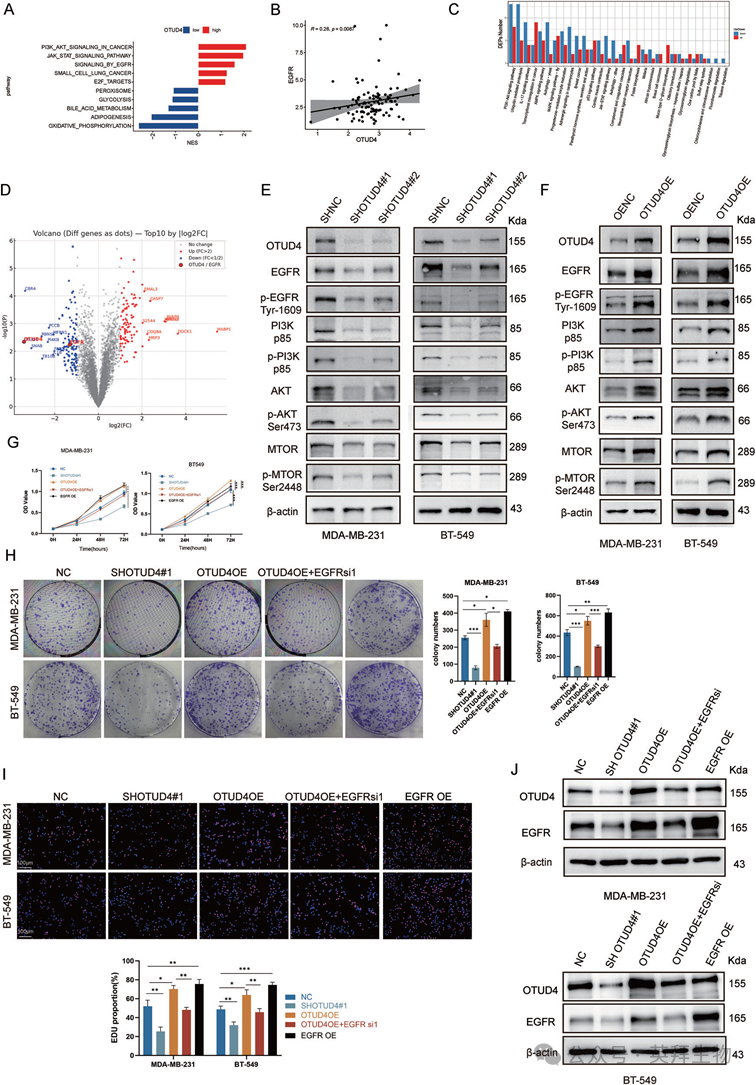
3)OTUD4在体外通过EGFR/PI3K/AKT通路促进TNBC的进展
为了阐明OTUD4介导TNBC侵袭的分子机制,我们进行了富集分析,发现OTUD4上调与TNBC中EGFR和PI3K/AKT通路之间存在强烈关联(图3A)。相关分析进一步证实OTUD4与EGFR表达呈显著正相关(图3B)。定量蛋白质组学分析显示,与对照组相比,MDA-MB-231细胞中OTUD4的敲低降低了OTUD4和EGFR蛋白水平(图3D)。KEGG富集分析表明,OTUD4沉默后PI3K/AKT通路下调(图3C)。此外,western blot结果显示,OTUD4敲除后,关键EGFR/PI3K/AKT通路组分的总磷酸化水平降低(图3E),而OTUD4过表达激活了该通路(图3F)。为了证实OTUD4通过EGFR发挥其致癌作用,我们进行了挽救实验。在OTUD4过表达的细胞中,EGFR的敲低消除了OTUD4过表达的促肿瘤作用(图3G-I)。在蛋白水平上,EGFR敲低抵消了OTUD4诱导的EGFR稳定,而OTUD4的稳定性不受EGFR敲低的影响(图3J)。这些结果表明OTUD4通过EGFR激活PI3K/AKT通路促进TNBC进展。
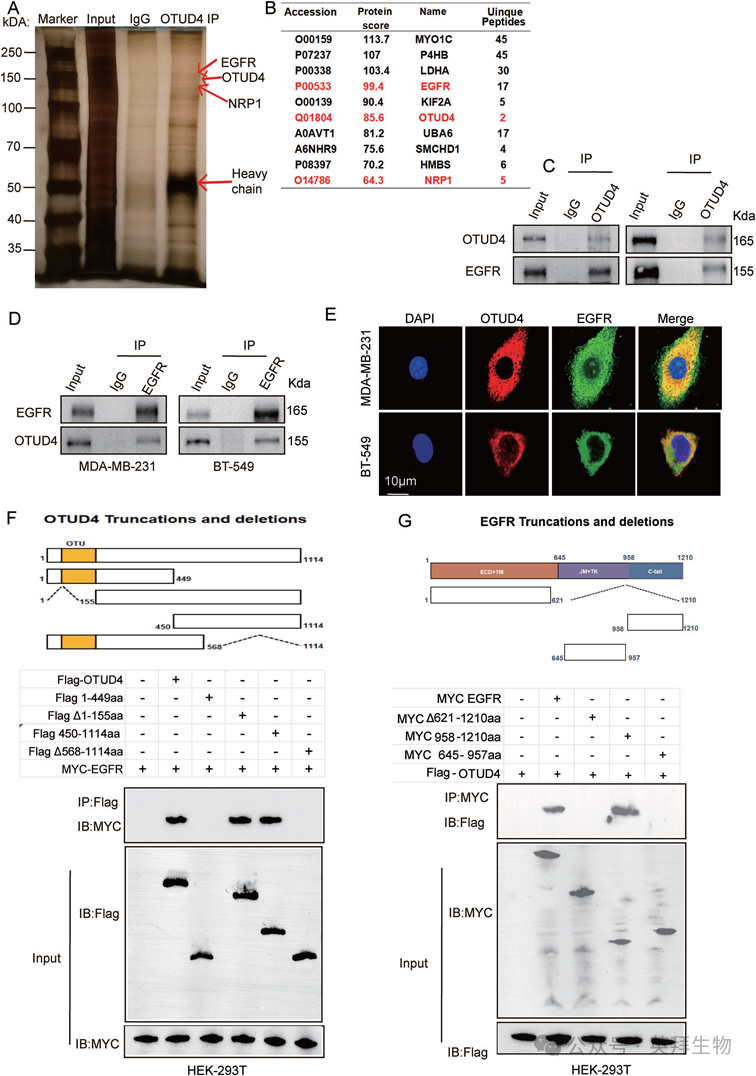
4)OTUD4和EGFR之间的相互作用是TNBC进展的驱动因素
为了鉴定EGFR/PI3K/ AKT通路中受OTUD4影响的靶基因,我们进行了免疫沉淀,然后进行了液相色谱-串联质谱(LC-MS/MS)鉴定OTUD4相互作用蛋白(图4A)。分析显示EGFR是OTUD4的潜在结合伙伴(图4B)。鉴于这种相互作用,我们假设OTUD4通过EGFR调节PI3K/AKT通路。共免疫沉淀实验证实了TNBC细胞中内源性OTUD4-EGFR相互作用(图4C,D),而IF分析显示OTUD4和EGFR共定位(图4E)。为了进一步描绘它们的特异性结合区域,我们构建了带有标记为OTUD4 Flag的截断和缺失变体的质粒:全长1- 1114aa,1-449aa, 450-1114aa,Δ1-155aa,Δ568-1114aa和myc标记的EGFR截断和缺失:1-1210aa,Δ621- 1210aa,958-1210aa,645-957aa。在HEK-293T细胞中转染后,随后的CO-IP分析证实了OTUD4 (568-1114aa)和EGFR (958- 1210aa)之间的相互作用
(图4F,G)。
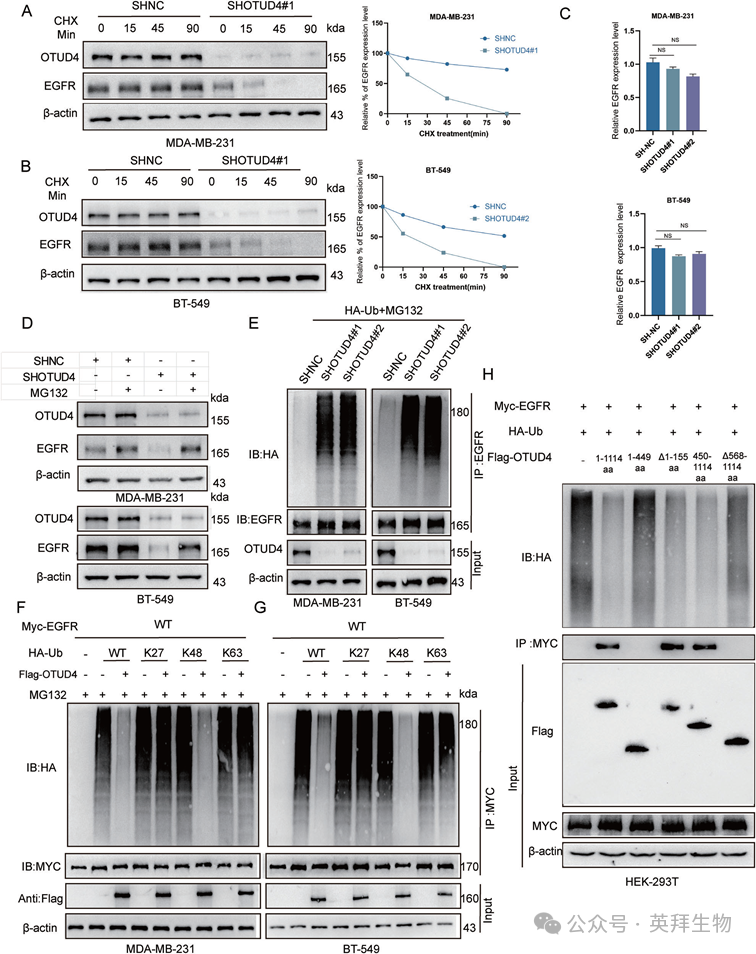
5)OTUD4通过去泛素化稳定EGFR表达
为了研究OTUD4调控EGFR表达的机制,我们考虑了它作为去泛素化酶的作用及其对EGFR蛋白稳定性的潜在影响。鉴于已报道的EGFR半衰期,我们进行了CHX追踪试验来评估EGFR随时间的降解。结果显示,OTUD4敲除显著降低了EGFR的半衰期(图5A,B)。然而,RT-qPCR分析证实,OTUD4敲低并未改变EGFR mRNA水平(图5C)。为了进一步探讨这一点,我们用蛋白酶体抑制剂MG132处理细胞,观察到MG132恢复了OTUD4-敲除细胞中的EGFR蛋白水平(图5D)。此外,泛素化实验表明MG32稳定的EGFP泛素化(图5E)。OTUD4过表达特异性地降低了k48连接的EGFR多泛素化,而不影响K63连接的泛素化(图5F,G)。为了确定EGFR去泛素化的功能域,我们使用OTUD4截断结构进行了泛素化分析。结果表明,全长OTUD4及其549-1114aa区可有效降低EGFR泛素化。我们推测568-1114aa区域对于OTUD4介导的EGFR去泛素化至关重要(图5H)。
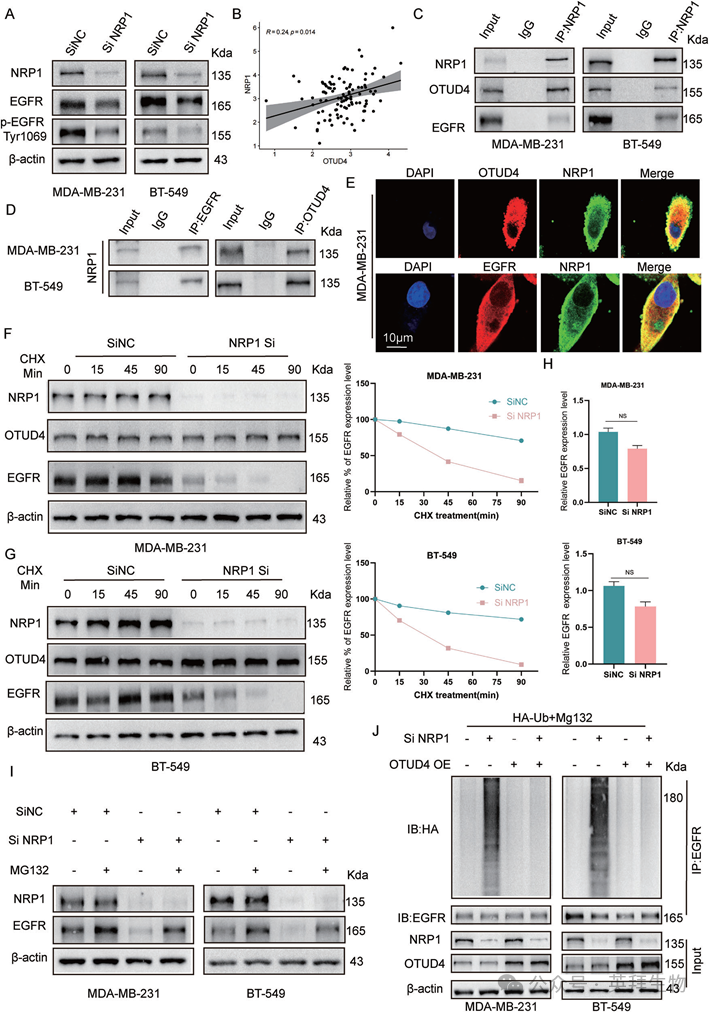
6)NRP1招募OTUD4去泛素化EGFR
质谱分析发现NRP1是潜在的OTUD4相互作用因子(图4A,B)。先前的研究报道了NRP1敲低可降低TNBC中的EGFR蛋白和磷酸化水平,但其潜在机制尚不清楚。我们证实了这一发现,表明TNBC细胞系中NRP1下调导致EGFR和p-EGFR蛋白水平下降(图6A)。相关分析进一步证实OTUD4与NRP1在TNBC中的表达呈正相关(图6B)。CO-IP实验证实了OTUD4和NRP1之间的相互作用(图4C,D,6C,D), IF分析显示它们在细胞质中共定位(图4E)。这些发现表明,OTUD4除了在去泛素化EGFR中的独立作用外,还可能被NRP1招募来稳定EGFR的表达。为了评估NRP1对EGFR稳定性的影响,我们进行了泛素化实验。CHX追逐实验显示,与对照组相比,NRP1-敲除TNBC细胞中EGFR的半衰期明显缩短,但不影响OTUD4的表达(图6F-G)。RT-qPCR分析证实NRP1在mRNA水平上不调节EGFR(图6H)。此外,MG132处理恢复了EGFR的稳定性(图6I)。免疫沉淀和Western blot分析进一步表明,OTUD4介导的NRP1依赖性去泛素化调节EGFR的稳定性(图6J)。这些结果表明NRP1招募OTUD4介导EGFR去泛素化。
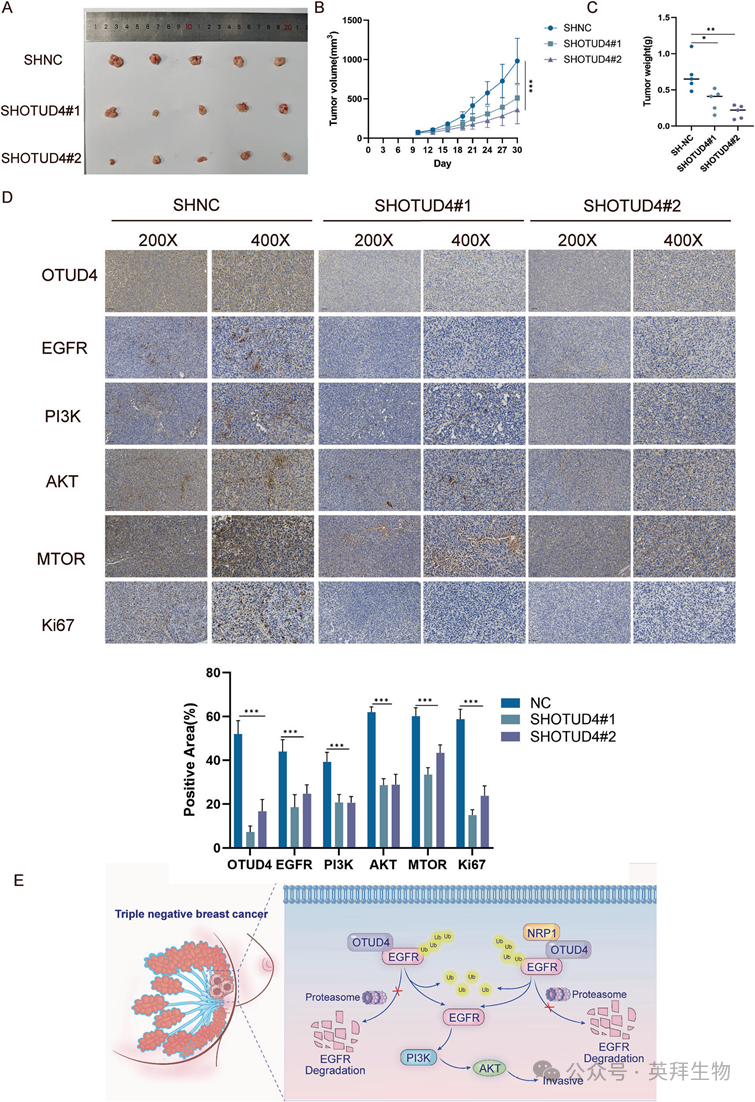
7)OTUD4通过EGFR/ PI3K/AKT通路促进TNBC在体内的增殖
为了评估OTUD4在TNBC体内增殖中的作用,我们将稳定的SHNC、SHOTUD4#1和SHOTUD4# 2 MDA-MB-231细胞皮下注射到雌性裸鼠腋下。肿瘤生长测量显示,OTUD4敲低显著抑制肿瘤进展(图7A-C)。免疫组织化学分析显示,与对照组相比,OTUD4敲除肿瘤中OTUD4、EGFR、PI3K、AKT、mTOR和Ki67的表达降低,表明OTUD4通过EGFR介导的PI3K/AKT途径促进TNBC肿瘤生长(图7D)。
结论:
OTUD4 在TNBC中过表达,并通过稳定EGFR增强其侵袭性,从而激活 PI3K/AKT 信号通路。OTUD4 通过直接去泛素化以及通过NRP1的招募来稳定 EGFR,为TNBC中EGFR的调控提供了新的见解。这些发现为未来针对OTUD4 的治疗策略提供了理论支持。
参考文献:
Ren Y, Zhou F, Tan Z, Yang S, Zhang S, Zhang Y, Fu Y, Zhang M, Liu S. OTUD4 deubiquitination stabilizes EGFR and activates the PI3K/AKT pathway to promote the invasiveness of triple-negative breast cancer. Cell Death Dis. 2026 Feb 23;17(1):245. doi: 10.1038/s41419-026-08482-x.