






新机制靶点!丹参素镁B通过干预GPX4-FUNDC1通路,对抗败血症肺损伤
败血症是一种危及生命的器官功能障碍,源于宿主免疫反应失调对严重感染。败血症相关肺损伤(SALI)是最常见且严重的表现之一,显著导致败血症患者高死亡率。早期有针对性地干预以减轻肺损伤及其后恶化,对于改善患者预后至关重要。肺微血管内皮损伤是SALI的早期标志,导致血管通透性增加和肺功能受损,使内皮成为阻止疾病进展的有力治疗靶点。
铁死亡是一种由铁依赖脂质过氧化驱动的调控细胞死亡形式,已成为影响败血症预后的关键病理过程。谷胱甘肽过氧化物酶4(GPX4)是一种将脂质过氧化物还原为相应醇的关键抗氧化酶,在抑制铁死亡中发挥关键作用。然而,SALI中GPX4蛋白水平显著下降,导致内皮对铁死亡、血管屏障破坏及随之而来的肺水肿敏感性增强。目前,SALI期间GPX4下调的机制尚不明确,且尚无有效干预措施能恢复GPX4水平和活性。线粒体吞噬是一种选择性自噬清除受损或功能失调线粒体的方法,对于维持细胞稳态以及减轻多种病理条件下的过度炎症和氧化应激至关重要。含FUN14结构域蛋白1(FUNDC1)是一种线粒体外膜蛋白,已被证明通过增强与LC3B的相互作用,促进线体吞噬,与败血症模型中的结局改善相关。新的证据显示,FUNDC1与GPX4之间存在有害相互作用,即FUNDC1将GPX4招募到线粒体,促进其通过线粒体的降解。因此,破坏GPX4-FUNDC1的相互作用,为减轻内皮损伤和改善SALI结局提供了有前景的方法。石胚酸钙B(MLB)是鼠尾草碱提取物中最丰富的生物活性成分,具有强效的抗氧化、线粒体保护和抗炎特性。以往研究强调了MLB在炎症和缺血再灌注引起的器官损伤中的保护作用。然而,MLB在SALI中的治疗潜力和潜在机制尚未明确。该研究2026年1月发表在《Advanced Science》,影响因子:14.1。
技术路线:

主要研究结果:
1、GPX4的内皮特异性缺失加重血管损伤和肺损伤
为观察感染条件下肺内皮中GPX4表达是否发生变化,作者对人类肺组织进行了免疫荧光染色。与未感染样本相比,感染肺部内皮细胞CD31GPX4显著减少,表明炎症内皮中的GPX4表达受抑制(图1A)。接受CLP治疗的小鼠肺组织CD31GPX4细胞数量也一致减少(图1B)。为进一步验证这些发现,作者重新分析了CLP手术后小鼠肺部的公开单细胞RNA测序数据(GSE207651)。UMAP分析确认了主要肺细胞群体的存在(图1C),并显示CLP处理小鼠肺组织内皮细胞中GPX4表达显著下降(图1D,E)。定量显示GPX4高CLP小鼠肺部内皮细胞相对于假对照组(图1F)。伪时间轨迹分析显示,GPX4表达高且铁死亡评分低的内皮细胞在轨迹根部富集,而GPX4降低且铁剥离子电位升高的细胞则出现在后期分支(图1G),表明在疾病进展过程中可能向铁死亡易感转变。
为功能性评估内皮GPX4在SALI中的作用,作者生成了内皮特异性Gpx4敲除小鼠(GPX4f/fCdh5Cre)使用Cre-LoxP系统(图1H)。在CLP的质疑下,GPX4f/fCdh5Cre与Gpx4相比,小鼠的生存率显著降低f/f小鼠(见图1I),表明内皮GPX4在败血症期间具有保护作用。肺血管通透性通过Evans蓝(EB)染料渗出和肺湿干重量比(W/D)评估,CLP小鼠显著升高,且在GPX4f/fCdh5Cre小鼠(图1J-L)。H&E染色显示,两种基因型的肺在假条件下表现出最小的改变。相比之下,CLP诱导肺泡壁增厚、炎症浸润和结构破坏,这些变化在GPX4f/fCdh5Cre老鼠。相应的肺损伤评分证实,内皮GPX4缺失时肺损伤显著加重(图1M,N)。综合来看,这些结果表明,在败血症期间,肺内皮细胞中的GPX4表达下调,其内皮特异性缺失加剧了血管通透性、炎症和肺损伤。
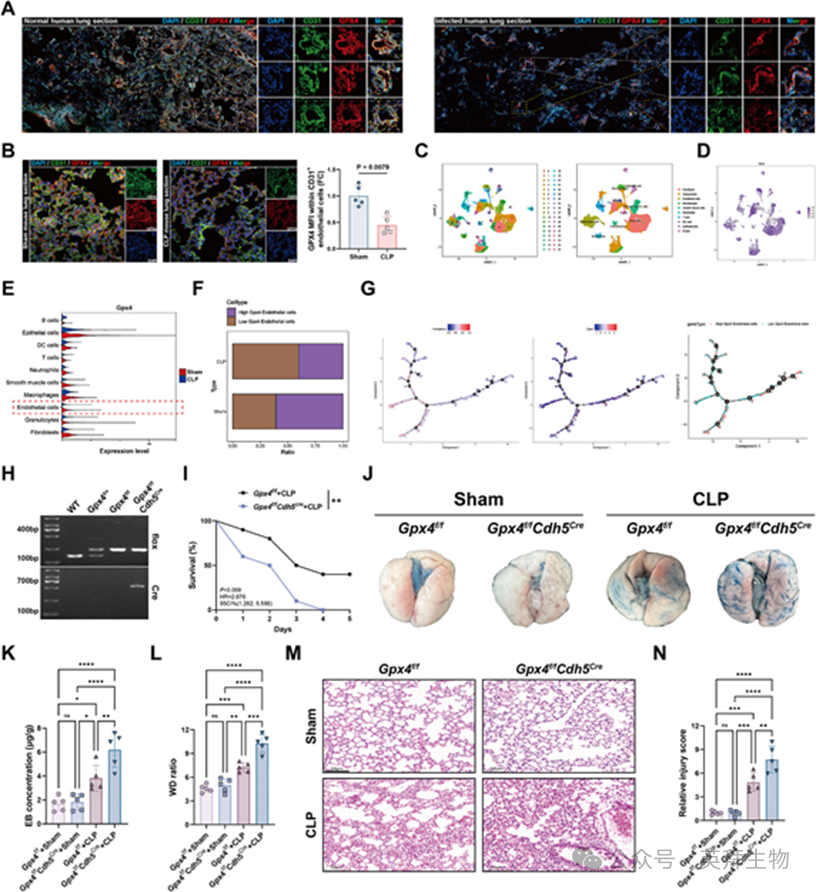
图1:内皮GPX4在败血症相关肺损伤中下调,而内皮特异性缺失则加重肺损伤
2、MLB直接结合GPX4,增强其酶活性,并减轻内皮细胞中的脂质过氧化
鉴于GPX4在抑制铁消亡和维持氧化还原稳态中的关键作用,作者进行了对天然产物库的计算机筛选,以识别潜在的GPX4靶向化合物。多步虚拟筛选流程包括HTVS、SP/XP对接和MM-GBSA重评分,最终产生了19个候选项目(图2A)。基于结合亲和力和已知的生物活性特征,选定了石本酸钙B(MLB)进行进一步评估(图2B)。为评估MLB在败血症期间的功能影响,作者使HPMECs接触来自LPS刺激巨噬细胞的调控培养基(CM)。C11-BODIPY染色显示CM处理细胞中脂质过氧化升高,MLB处理显著减弱(图2C),表明其抗氧化潜力。为确定MLB是否能直接抵消GPX4抑制诱导的铁凋亡,作者用RSL3处理了HPMEC。MLB显著减少了RSL3诱导的细胞死亡和脂质过氧化(见图2D-G),强化了其铁灭亡抑制剂的作用。功能上,MLB剂量依赖性增强GPX4酶活性,并部分挽救了RSL3对其抑制作用(图2H)。为了探索原子层面的势相互作用机制,作者利用密度泛函理论(DFT)进行了静电势(ESP)分析。球棒和ESP映射显示MLB空间定位于Sec46含硒侧链附近(图2I)。值得注意的是,MLB上的负电位区域与亲电硒中心对齐,暗示可能存在弱轨道相互作用,可能有助于GPX4活性的变构调控。
接着作者调查了MLB是否直接调节GPX4。分子对接显示出稳定的MLB-GPX4复合物,预测结合自由能为−8.89 kcal/mol(图2J)。分子动力学模拟一致确认了稳定结合,显示约10纳秒时收敛,RMSD稳定持续100纳秒(图2K)。均方根涨落(RMSF)分析进一步显示,关键绑定区,特别是GPX4残基75和135附近,原子波动较低(图2L)。为了验证细胞中的靶点结合,作者进行了细胞热移分析(CETSA)。温移分析显示,MLB预处理提高了内源性GPX4的热稳定性(图2M)。52°C单温度CETSA进一步显示稳定GPX4在12.5-200μ mMLB范围内剂量依赖性增加(图2N)。表面等离激元共振(SPR)进一步验证了MLB与重组GPX4之间的直接相互作用,解离常数为25.94μm(图2O)。综合来看,这些结果表明MLB直接结合GPX4,增强其酶功能,并减轻内皮细胞中的脂质过氧化,支持其作为GPX4靶向铁死亡调节剂的潜力。
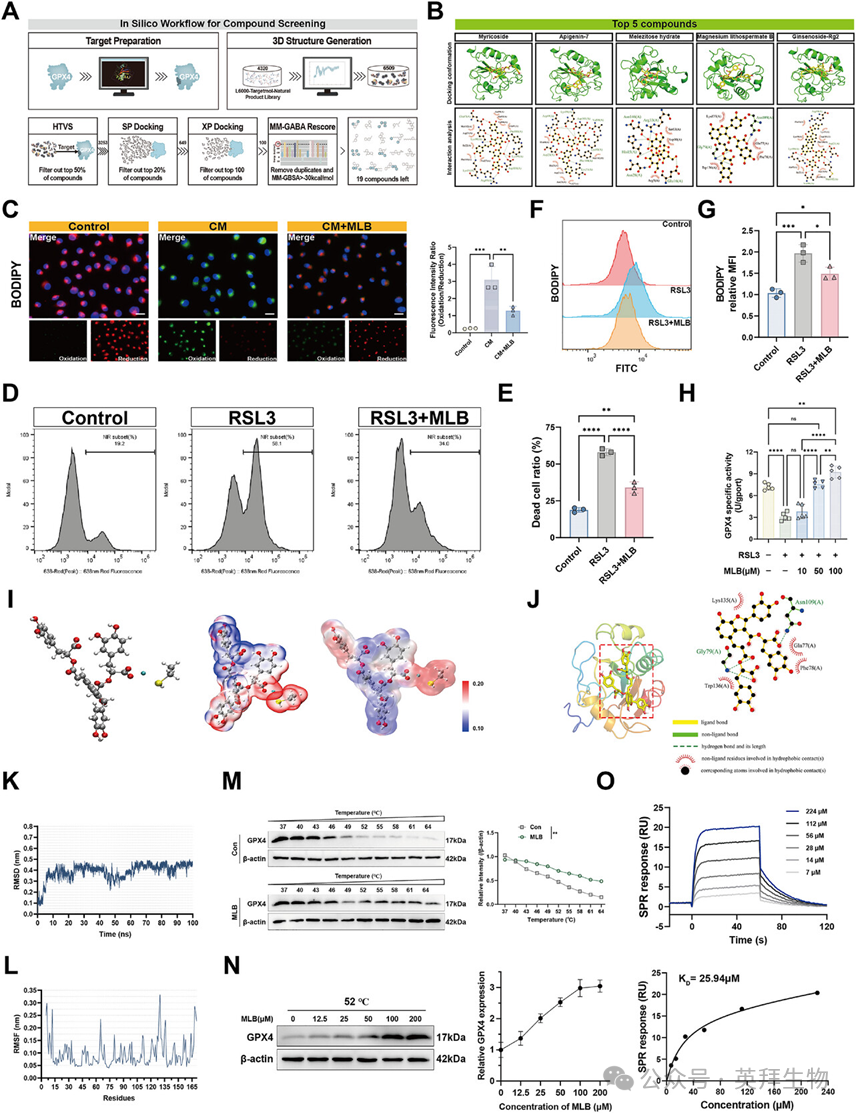
图2:MLB直接结合GPX4,增强其酶活性,并减少HPMEC中的脂质过氧化。
3、MLB缓解败血症期间内皮细胞的铁消亡和屏障功能障碍
为进一步探讨MLB的保护作用,作者通过用LPS刺激THP-1来源巨噬细胞并收集其CM以治疗HPMECs,建立了体外败血症模型(图3A)。细胞毒性分析显示,高浓度的MLB(500μm)降低了细胞存活能力,而100μm则耐受良好(图3B)。值得注意的是,MLB显著恢复了CM暴露所抑制的细胞存活能力(图3C)。流式细胞术分析显示,CM处理的HPMEC细胞死亡增加,表现为僵尸近红外阳性升高。该效应被MLB或铁消亡抑制剂铁抑素-1(Fer-1)有效逆转,且两者无显著差异(图3D)。西方印迹和免疫荧光分析显示,CM显著下调紧密连接蛋白ZO-1、VE-Cadherin和occludin,表明内皮屏障破坏。MLB和Fer-1部分恢复了这些标志物的表达(图3E,F),表明MLB能减轻CM诱导的内皮屏障障碍障碍。氧化还原状态评估显示,CM使GSSG和MDA水平升高,而GSH降低,符合氧化应激增加的表现。这些变化通过MLB或Fer-1处理得到改善(图3G)。此外,CM显著抑制关键铁剥离调控因子SLC7A11和GPX4的表达,这两者均通过免疫印迹和免疫荧光显示,均被MLB或Fer-1部分挽救(图3H,I)。为进一步验证脂质过氧化,作者进行了Liperfluo染色和C11-BODIPY染色。两项检测均确认CM处理细胞中脂质ROS显著增加,且MLB或Fer-1可逆转(图3J,K)。总体证据表明,MLB通过调节GPX4依赖的抗氧化物防御,有效减弱CM诱导的铁坏死和内皮功能障碍。
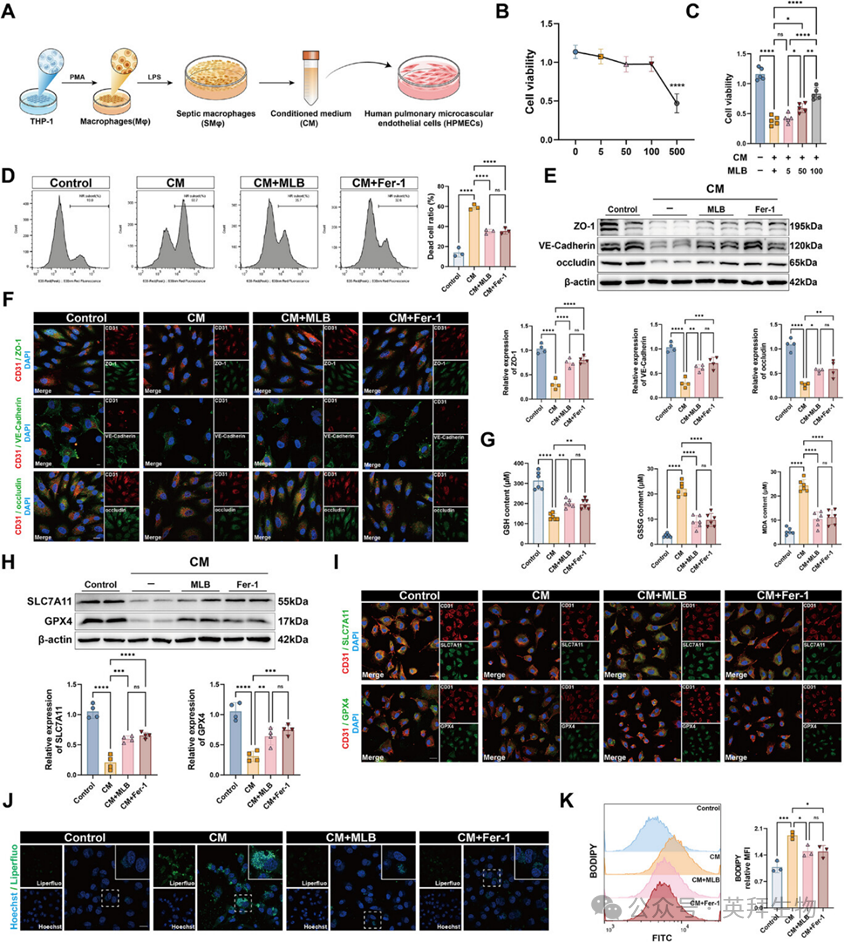
图3:MLB能减轻HPMEC中败血症诱导的铁消亡和屏障功能障碍。
4、MLB通过GPX4依赖机制保护肺血管内皮细胞免受败血症诱发的铁死亡和屏障破坏
为了确保MLB在生理条件下的生物安全,作者首先评估了其在健康小鼠中诱发炎症的潜力。接着作者考察了MLB对CLP治疗小鼠肺部铁磷遁症的影响。基于上述发现,作者选择了高剂量MLB作为后续实验的30mg/kg。TUNEL和DHE染色显示CLP小鼠肺部中死细胞和ROS阳性细胞显著增加,MLB处理显著减少了这一现象(图4A,B)。MLB还逆转了CLP引起的氧化还原失衡,表现为GSSG和MDA水平降低以及GSH水平升高(图4C)。免疫荧光和免疫印迹显示,GPX4和SLC7A11表达在CLP小鼠肺部被下调,MLB部分恢复(图4D,E)。透射电子显微镜显示CLP小鼠肺部线粒体收缩和膜密度增加,MLB缓解了这些现象(图4F),支持了超微结构层面铁死亡的抑制。
为评估MLB的保护效果是否依赖于内皮GPX4,作者比较了以下反应GPX4f/f以及GPX4f/fCdh5Cre小鼠在CLP诱导后接受MLB治疗。与 GPX4 比较f/f老鼠们,GPX4f/fCdh5Cre小鼠表现出显著增加的埃文斯蓝渗出现象(图4G),肺湿干比例升高(图4H),以及血清中Angpt2水平较高(图4I)。组织学检测进一步显示肺部损伤加重且ROS积累增强GPX4f/fCdh5Cre尽管有MLB管理(图4J,K)。这些发现表明,MLB主要通过依赖GPX4的机制对肺血管屏障发挥保护作用。
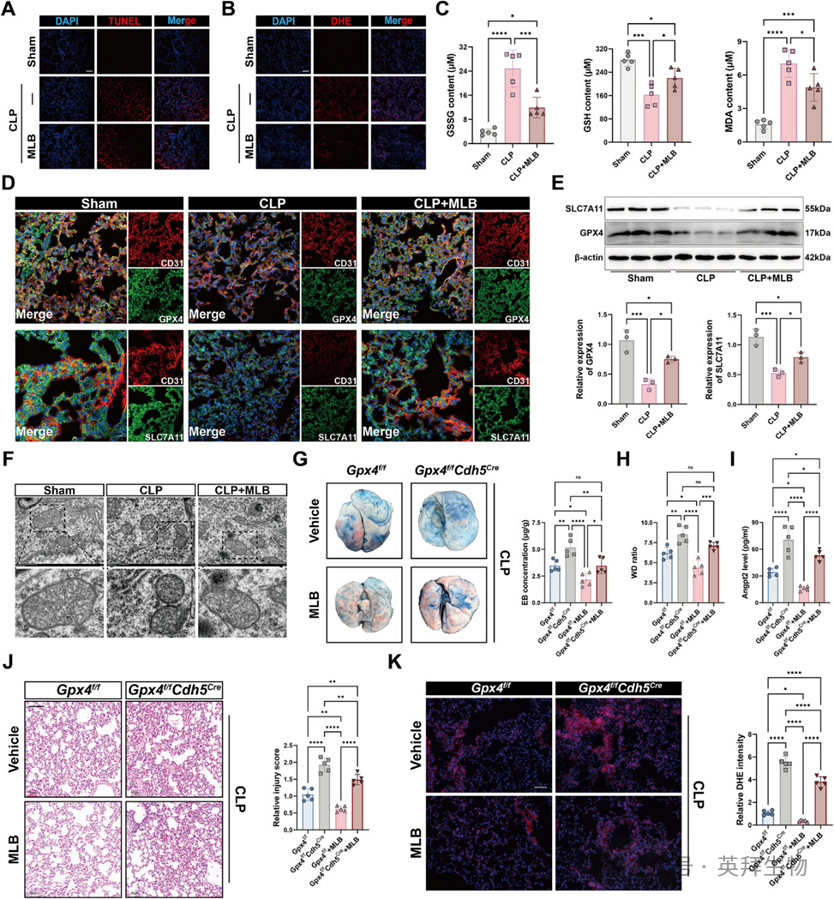
图4:MLB通过上调GPX4来缓解CLP诱导败血性小鼠的肺铁死亡。
5、MLB稳定GPX4并维持CM处理HPMEC中的线粒体稳态
为阐明MLB上调GPX4表达的机制,作者研究了GPX4在被确认结合MLB的翻译后调控作用。在CM条件下,单独使用MG132、BafA1或氯喹(CQ)治疗部分恢复GPX4蛋白水平。与MLB和MG132联合治疗相比单独治疗,GPX4水平进一步升高。相比之下,MLB与BafA1或CQ联合使用,并未带来超出MLB或溶酶体抑制单独实现的额外上调(图5A,B)。综合来看,这些结果表明MLB至少部分通过自噬溶酶体途径调节GPX4的稳定性,而非直接阻断蛋白酶体降解。
此外,作者重新分析了CLP小鼠肺部的单细胞RNA测序(scRNA-seq)数据,重点关注肺内皮细胞。KEGG途径对不同表达基因的富集GPX4高以及GPX4低细胞显示线粒体相关通路富集,暗示GPX4与线粒体稳态之间存在潜在联系。先前研究表明,FUNDC1会招募GPX4到线粒体,线粒体促进其降解并促进铁凋亡。基于这些发现,作者推测MLB诱导的GPX4上调可能与其对线粒体作用机制相关。
为验证该假设,作者评估了HPMECs中的线粒体完整性和线粒体吞噬活性。MitoTracker Red染色显示,对照组线粒体表现出丝状形态,而CM治疗则导致明显的线粒体破碎和萎缩。MLB给药部分恢复了正常的线粒体形态(图5C)。线粒碱和JC-1染色一致显示CM提高了线粒体ROS水平并降低膜电位(ΔΨm),这两者均被MLB处理显著逆转(图5D-G)。西方印迹分析进一步显示MLB下调了TOM20,同时上调了自噬标志物LC3B(图5H)。免疫荧光染色显示,MLB处理组中TOM20与LC3B的共定位增强,相较于单用CM组(图5I),与线粒体吞噬活性增强相符。综合来看,这些结果表明MLB促进线粒体吞噬,并在败血症期间维持肺内皮细胞的线粒体稳态。
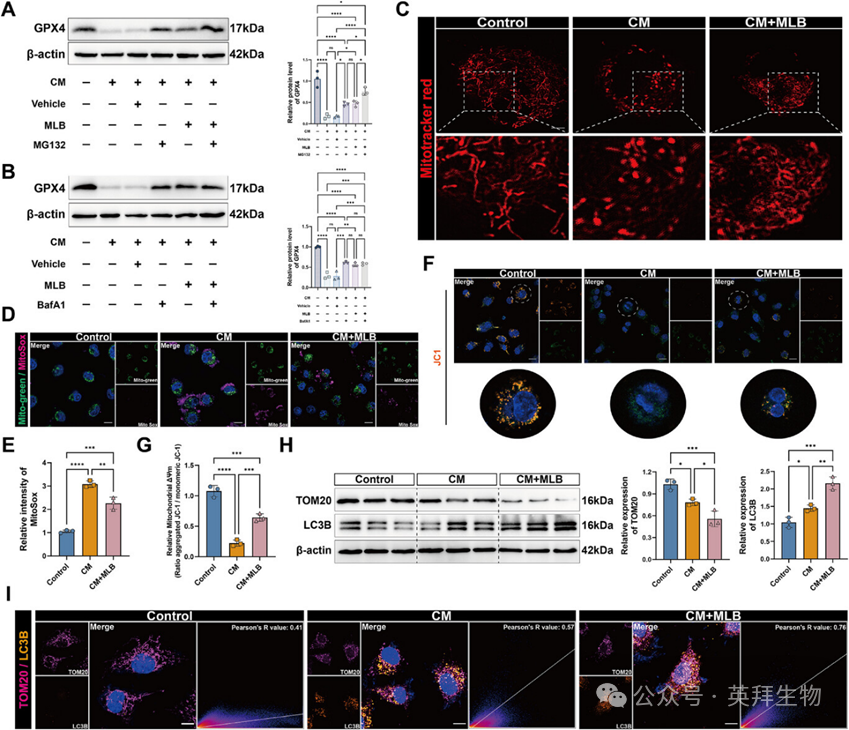
图5:MLB恢复CM处理HPMECs中的线粒体功能并增强线粒体吞噬能力。
6、MLB通过线粒吸收破坏GPX4-FUNDC1的相互作用并缓解GPX4降解
先前研究表明,FUNDC1与GPX4相互作用,这不仅损害FUNDC1与LC3B的结合,从而降低线体吞噬效率,还促进GPX4的线粒体易位,从而通过线粒吞噬途径促进其降解。作者的初步数据表明,MLB通过抑制溶酶体依赖的降解,同时增强线粒吞噬活性并提高GPX4蛋白水平,促使MLB可能通过破坏GPX4-FUNDC1相互作用来发挥保护作用的假说。为验证这一假设,作者进行了蛋白质-蛋白质连接和分子动力学模拟,以评估GPX4-FUNDC1复合物的稳定性。GPX4与FUNDC1之间的结合界面涉及多重静电和氢键相互作用(图6A)。RMSD结果显示,GPX4-FUNDC1复合物在约10纳秒后达到结构稳定性(图6B)。RMSF分析进一步揭示GPX4残基75周围区域的柔韧性降低(图6C)。这些结果支持GPX4与FUNDC1之间存在稳定相互作用。
共免疫沉淀测定进一步证实,CM处理增强了GPX4-FUNDC1复合物的形成,而MLB显著破坏了该相互作用,同时促进FUNDC1-LC3B结合(图6D)。共焦点免疫荧光成像显示,CM随时间促进GPX4-LC3B共定位,而MLB治疗则逆转这一趋势,反而增强了LC3B-MitoTracker共定位,这与线粒体吸收增加和GPX4线粒体降解减少相符(图6E)。这些结果表明MLB破坏了GPX4-FUNDC1复合物,从而促进FUNDC1介导的线粒体吞噬,缓解GPX4的线粒吞噬降解。
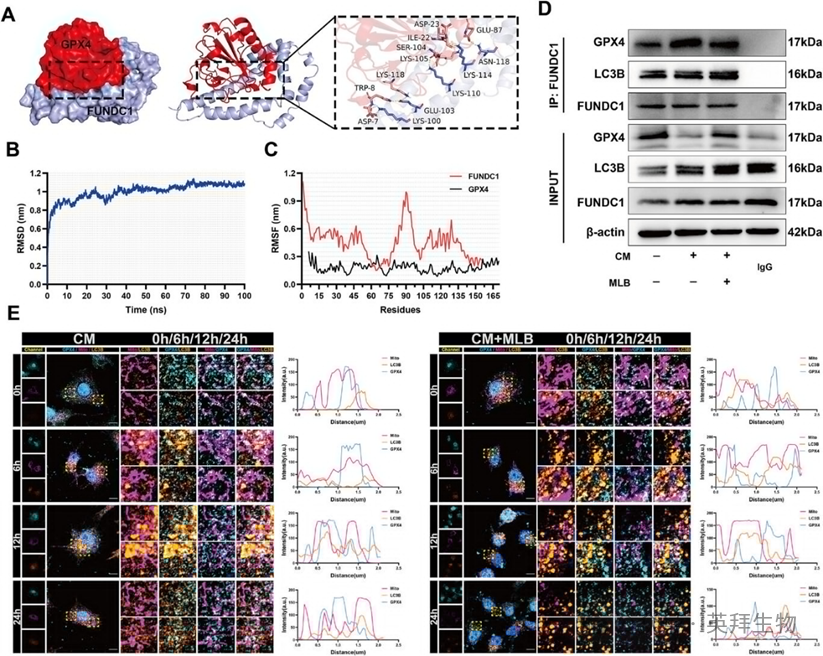
图6:MLB破坏GPX4-FUNDC1的相互作用,促进FUNDC1-LC3B结合。
7、MLB靶向GPX4的Gly79,破坏GPX4-FUNDC1的相互作用,从而在败血症中保持内皮完整性和线粒体功能
分子对接分析鉴定了GPX4的Gly79和Asn109为MLB潜在的氢键残基。为验证其作用,作者生成了单点突变体G79S(甘氨酸变丝氨酸)和N109S(天冬氨酸转丝氨酸)。编码野生型GPX4(GPX4-WT)、GPX4-G79S或GPX4-N109S的质粒被转染到HPMEC中以进行功能表征。共免疫沉淀显示MLB破坏了GPX4-WT和GPX4-N109S细胞中的GPX4-FUNDC1相互作用,而该效应在GPX4-G79S细胞中明显减弱(图7A)。为直接评估Gly79对MLB结合的贡献,作者纯化了重组GPX4-WT和GPX4-G79S蛋白,并进行了差分扫描荧光测量(DSF)。MLB显著提升了GPX4-WT的热稳定性,但对GPX4-G79S的稳定作用有限。这些检测结果共同表明,Gly79对MLB-GPX4的结合至关重要,是MLB介导的GPX4-FUNDC1复合物破坏的基础。
功能上,MLB未能逆转CM引起的MDA和GSSG水平升高,也未能恢复GPX4-G79S细胞中的GSH含量(图7B)。ROS染色和MitoTracker检测显示,MLB在GPX4-WT细胞中降低了氧化应激并保留了线粒体形态,而这些效应在G79S突变体中被消除(图7C,D)。同样,流式细胞术显示MLB显著减少了GPX4-WT细胞中CM诱导的细胞死亡,但在G79S细胞中保护作用极低(图7E)。在GPX4-G79S转染的HPMEC中,MLB也未能恢复CM诱导的紧密连接蛋白和铁剥离调控因子的减少(图7F,G)。酶活性检测进一步证实,G79S突变显著削弱了MLB对GPX4活性的修复作用(图7H)。
CLP小鼠通过尾静脉注射编码GPX4-WT或GPX4-G79S的AAV载体,随后进行MLB处理。H&E染色显示,MLB治疗显著减轻了GPX4-WT小鼠的肺损伤,但在GPX4-G79S小鼠中则无显著减弱,这一点从后者的损伤评分较高可见一斑(图7I)。埃文斯蓝渗出和DHE染色显示GPX4-G79S小鼠肺部血管通透性和ROS积累较GPX4-WT更高(图7I)。Western blot 分析证实,MLB诱导的屏障蛋白和铁坏死抑制因子在GPX4-G79S小鼠中显著减弱(图7J,K)。免疫荧光染色显示GPX4-G79S小鼠肺部GPX4和CD31共定位减少(图7L)。氧化应激标志物的定量进一步表明,MLB显著降低了GPX4-WT肺部的MDA和GSSG水平,并增加了GSH含量,但这些效应在G79S突变体中被消除(图7M)。综合来看,这些发现确立了GPX4的Gly79是MLB结合和治疗疗效的关键决定因素。
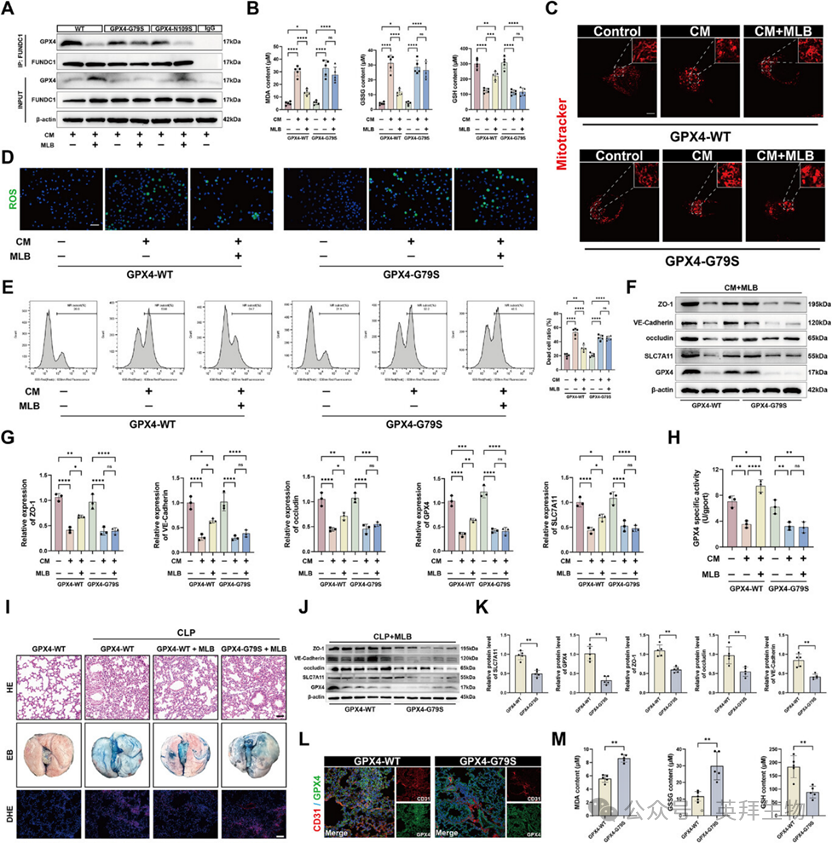
图7:MLB在Gly79与GPX4相互作用,破坏GPX4-FUNDC1的相互作用,减轻细胞损伤和线粒体功能障碍。
8、MLB与MitoQ联合治疗对SALI具有协同保护作用
鉴于MLB同时靶向GPX4和线粒体功能,作者研究了与线粒体靶向抗氧化剂米托册甲基酸盐(MitoQ)联合给药——该药物目前正临床评估用于内皮保护和线粒体保存——是否能提升治疗效果。为定量评估MLB与MitoQ的相互作用,作者在CM处理的HPMECs中采用CCK-8可行性分析,进行了6×6剂量反应矩阵。单纯增加任一药物浓度均能适度提升细胞存活率,而联合治疗在大多数剂量组合中均优于单药,效果最显著的表现为25-50m MLB加0.25-0.5m MitoQ μμm。Kaplan-Meier生存分析显示,虽然MLB或MitoQ单独在CLP手术后生存率略有提升,但两者联合使用相比单用治疗显著延长了5天生存期(图8A)。组织学检查显示,每种药物均部分缓解了CLP引起的肺损伤,联合治疗进一步减少了组织损伤,肺损伤评分降低(图8B,C)。DHE染色显示,结合组相较单剂治疗的ROS清除率更高(图8D)。MLB-MitoQ联合治疗还显著抑制肺部促炎细胞因子的水平,包括TNF-α、IL-6和IL-1β(图8E)。同样,氧化应激标志物反映了氧化还原稳态的改善:脂质过氧化(MDA)减少,GSH/GSSG平衡在联合治疗下更有效地恢复(图8F)。值得注意的是,MLB+MitoQ提供了更优越的血管屏障保护,表现为埃文斯蓝渗出减少和改善肺部W/D比,表明肺水肿减轻(图8G,H)。综合来看,这些发现表明MLB和MitoQ协同作用以减轻SALI,但其分子基础尚待澄清。
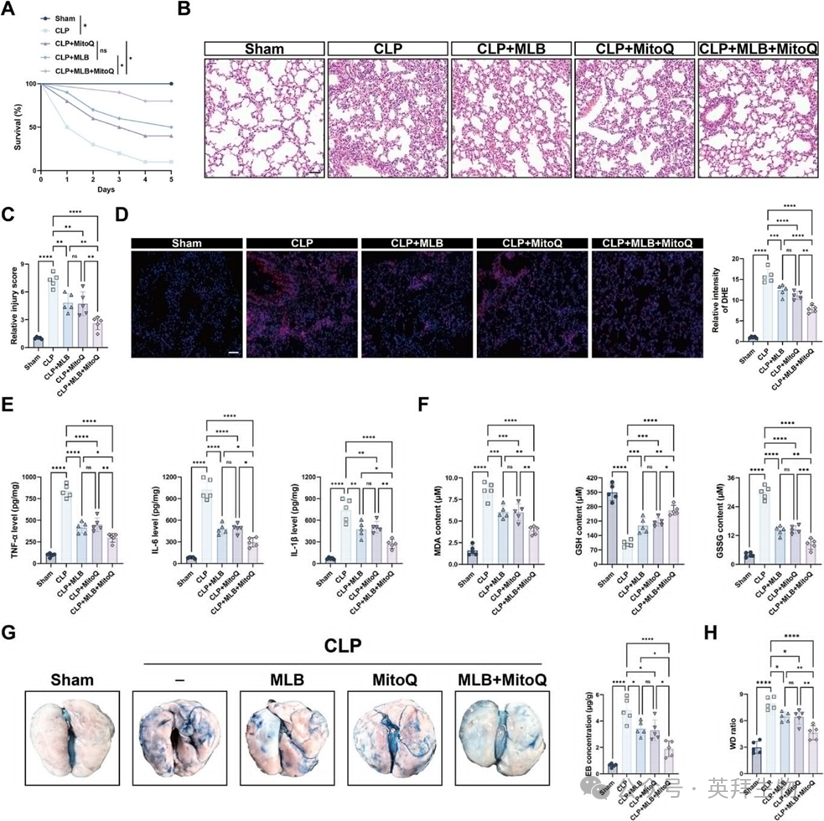
图8:与MLB和MitoQ联合治疗在败血性小鼠中提供更优越的肺部保护。
9、MLB@PBP-ADSC-EV增强肺部靶向,增强败血症中的肺部保护
为了提升MLB在SALI中的治疗效果,作者开发了一种基于脂肪来源干细胞(ADSC)的工程化细胞外囊泡(EV)的靶向递送系统。P-选择蛋白在受伤后(包括败血症)在肺血管内皮细胞上迅速上调。接着,作者从小鼠脂肪组织中分离并鉴定了ADSCs。为实现MLB输送,ADSC-EV与MLB一起孵育并接受超声辅助加载,实现了42.16%的封装效率。同时,P-选择素结合肽(PBP,CDAEWVDVS)与DMPE-PEG-MAL共价结合,生成DMPE-PEG-PBP(DPP),该结果通过马来酰亚胺峰(6.8 ppm)消失及PBP相关质子信号(7-7.6 ppm)出现得到证实。DPP入EV膜,Cy5.5荧光团与PBP结合进行成像(图9A)。最终产生的MLB@PBP-ADSC-EV保持了原生电动车的结构完整性和标志性特征,并且在Zeta势和颗粒尺寸上表现相当(图9B-E)。生物分布分析显示,静脉注射的Cy5.5标记MLB@PBP-ADSC-EV在注射后6至24小时内在肺部的积累比MLB@ADSC-EV更为稳健(CLP小鼠)(6至10×12颗粒数/公斤)。重要的是,24小时对主要器官的定量分析进一步显示,与MLB@ADSC-EVs相比,MLB@PBP-ADSC-EVs肺部信号增强更为明显,肝脏、脾脏和肾脏的摄取也增加,心脏无明显增多(图9F)。流式细胞术确认MLB@PBP-ADSC-EVs在4°C下至少保持结构稳定性7天,MLB释放动力学显示48小时内累计释放率达85%(图9I)。免疫荧光确认MLB@PBP-ADSC-EVs与肺内皮标志物CD31的共定位增强(图9J),而体外摄取测定显示CM处理的HPMEC内化增强。这些结果共同支持EVs的物理化学特性得以保存,MLB的高效装卸,以及MLB@PBP-ADSC-EV在肺内皮靶向的增强。
为评估MLB的肺部输送效率,作者采用基于SERS的策略量化MLB在肺组织和血清中的分布。SERS是一种高度灵敏且可重复的技术,适用于微量化合物检测。利用已有的方案,作者成功合成了Ag@CIT SERS底物(图10A)。有限差分时域(FDTD)模拟确认Ag@CIT表面存在电磁热点,有助于信号增强(图10B)。Ag@CIT基底大幅放大了MLB的拉曼信号,并实现了其特征频谱指纹的稳健获取(图10C),并实现了跨复制体的稳定读数。接着,作者加强了该SERS平台的定量表现。基于强度比构建了校准曲线 I1265/我2078其中以氘甲醇(DM)作为内部标准,适用于1.5625至50 μg/mL(R)范围内的MLB浓度2= 0.999)。基于标准方法(LOD = 3.3σ/k;LOQ = 10σ/k),该方法的检测灵敏度计算为LOD = 40.9ng/mL,LOQ = 136.6 ng/mL(图10D)。为评估潜在基质干扰,作者采集了空白小鼠血清和肺均质物的背景拉曼光谱;两个样本均未显示典型的MLB峰值,表明诊断区内内源性光谱干扰极小(图10E,F)。为提供正交验证,作者建立了MLB(R)高性能液相色谱-质谱(HPLC-MS)校准曲线2= 0.9978)以及小鼠血清中量化的MLB水平。六只小鼠的配对比较显示,SERS与HPLC-MS测量高度一致(Pearson R = 0.945,p = 0.004),支持基于SERS的体内定量的准确性和可靠性。作者定量评估静脉注射后肺部和血清中的MLB水平。值得注意的是,使用MLB@PBP-ADSC-EVs治疗的小鼠在肺组织中MLB浓度显著升高,血清水平降低,相较于游离MLB或非靶向MLB@ADSC-EVs,显示肺部输送效率有所提升。
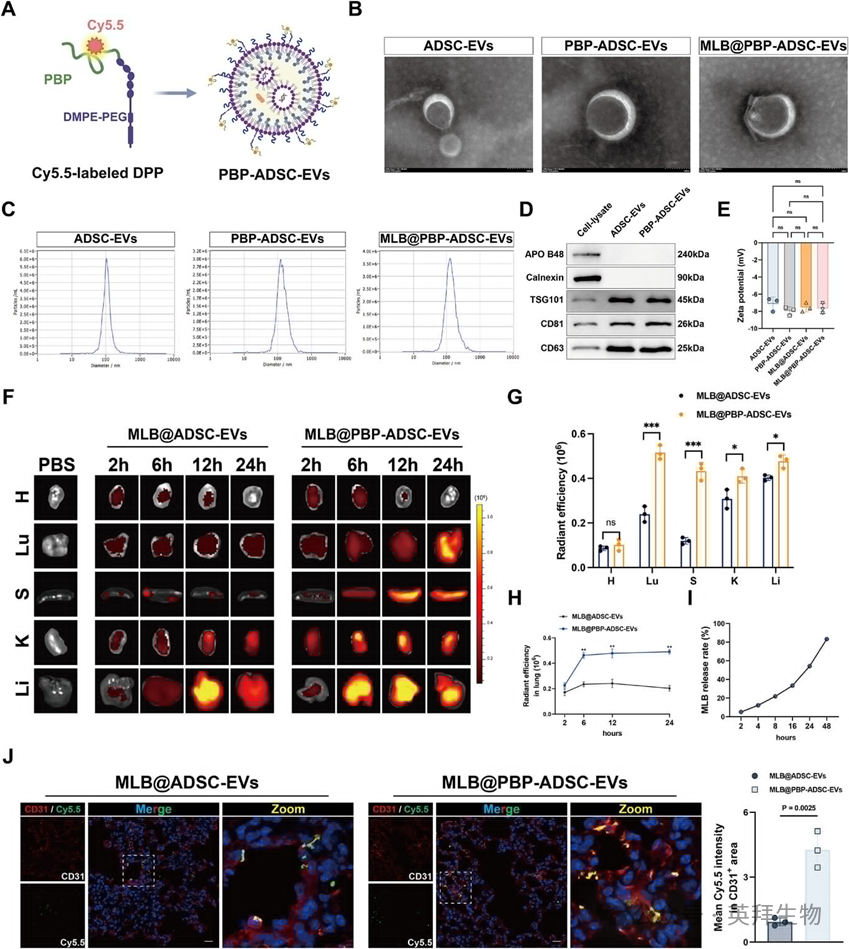
图9:靶向MLB@PBP-ADSC-EVs向化脓性小鼠发炎的肺内皮递送。
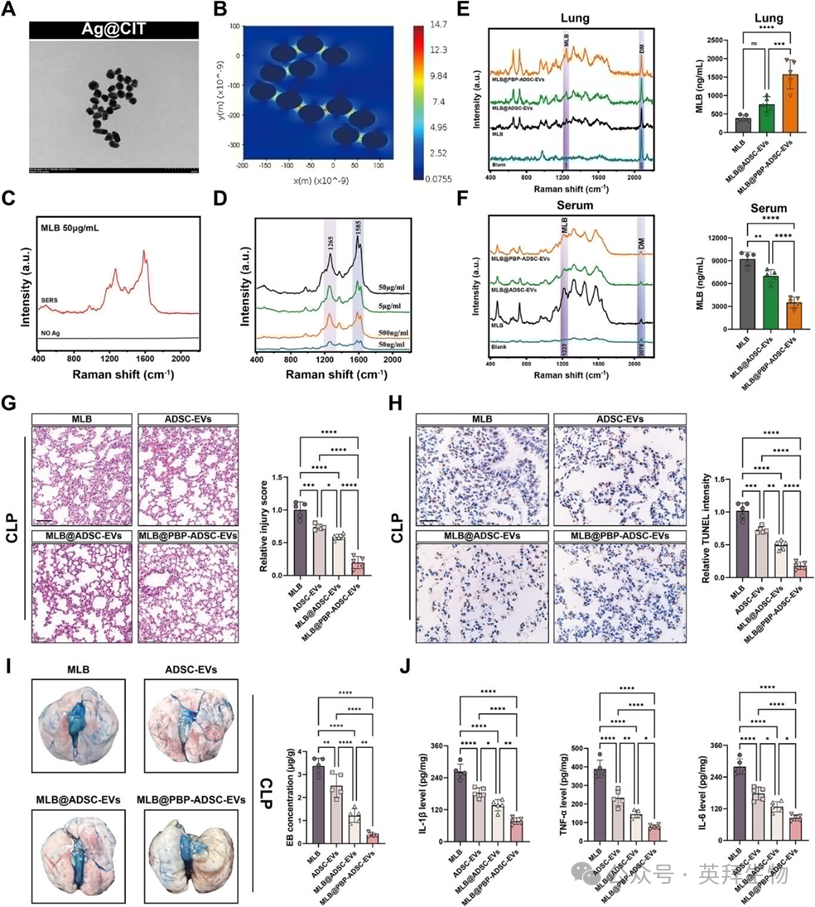
图10:MLB@PBP-ADSC-EV增强了CLP麦克风诱导化脓性小鼠的肺部保护。
接着作者研究了MLB@PBP-ADSC-EVs在SALI中的治疗效果。在CLP小鼠中,作者比较了MLB、ADSC-EV、MLB@ADSC-EV和MLB@PBP-ADSC-EVs,且所有含MLB组的MLB剂量相同,且基于基于电子运动的配方中EV数值相等。MLB@PBP-ADSC-EVs治疗显著减轻肺损伤,表现为肺损伤评分降低和TUNEL阳性细胞减少,且在所有治疗组中保护力最高,优于MLB、ADSC-EV和MLB@ADSC-EVs(图10G,H)。一贯地,MLB@PBP-ADSC-EVs最有效地降低肺组织中的血管通透性和促炎细胞因子水平(IL-1β、TNF-α和IL-6)(图10I,J)并抑制了ROS积累,如DHE荧光减弱。综合来看,这些结果表明MLB@PBP-ADSC-EV在SALI中比游离MLB和非靶向EV制剂提供更优越的肺保护。图11展示了MLB调控SALI内皮铁凋亡的拟定机制,并概述了靶向递送和分子检测的策略。
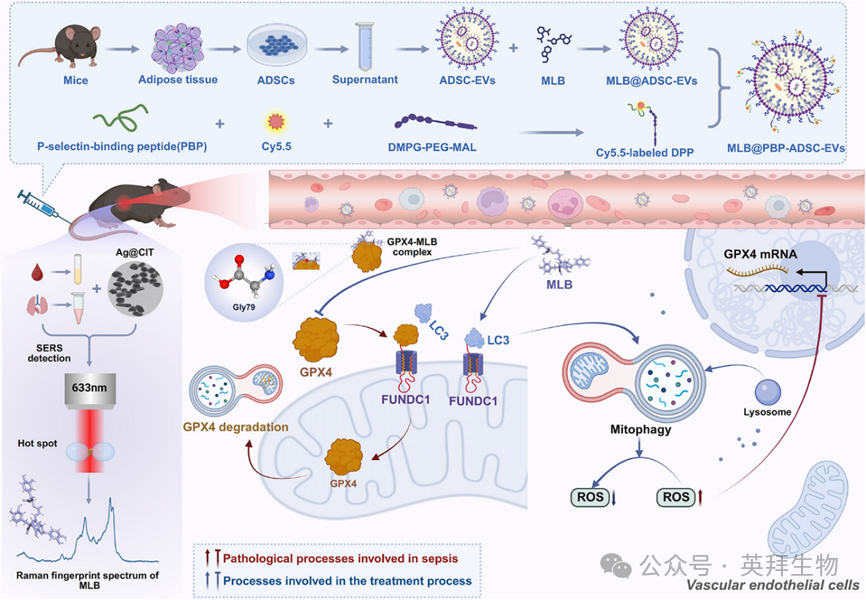
图11:示意模型展示了MLB的内皮保护机制及其在靶向递送和体内检测中的转化潜力。
结论:
该研究确认了天然小分子MLB作为GPX4的新型调节因子,能够破坏其与FUNDC1的相互作用,防止线粒体吞噬依赖性的GPX4降解,从而恢复FUNDC1介导的线粒吞噬通量,并减轻败血症期间的内皮铁坏死。此外,通过工程化MLB@PBP-ADSC-EV,作者实现了MLB对炎症内皮的靶向递送,显著提升了其在SALI中的治疗效果。此外,作者应用基于SERS的分子检测技术,实现体内MLB分布的定量监测。这些发现不仅澄清了MLB在SALI中的保护机制,还建立了支持其临床转化的靶向递送和检测策略。
参考文献:
Li Z, Liu C, Ma Z, Zheng D, Wang R, Yu Y, et al. Targeting the GPX4-FUNDC1 Interaction with Magnesium Lithospermate B Attenuates Sepsis-Associated Lung Injury. Adv Sci (Weinh). 2026 Jan 30:e16488. doi: 10.1002/advs.202516488. Epub ahead of print. PMID: 41616122.