






组蛋白甲基转移酶ASH1L 引发骨生态位中巨噬细胞的转移和代谢重编程
骨转移是癌症死亡的主要原因,然而驱动这一过程的表观遗传学决定因素仍不明确。本研究发现,组蛋白甲基转移酶ASH1L在男性前列腺癌患者中存在基因扩增现象,并且是骨转移过程的关键调控因子。ASH1L通过重塑组蛋白甲基化修饰,与HIF-1α协同作用诱导侵袭性癌细胞形成促转移转录组,导致单核细胞分化为脂质相关巨噬细胞(LA-TAM),并增强这些细胞在转移性骨微环境中的促肿瘤表型。我们鉴定出IGF-2作为ASH1L/HIF-1α的直接靶标,通过重编程氧化磷酸化过程介导LA-TAM的分化与表型改变。在临床前模型中,药物抑制ASH1L-HIF-1α-巨噬细胞轴可引发显著的抗转移反应。本研究揭示了癌细胞中的表观遗传学改变通过重编程代谢机制调控髓系细胞特征,从而促进转移灶生长的分子机制,确立了ASH1L作为表观遗传驱动因子在骨微环境中启动转移和巨噬细胞可塑性的关键作用,为转移性恶性肿瘤提供了可靠的治疗靶点。该研究于2025年5月发表在《Nature Communications》,IF:15.7。
技术路线:
主要研究结果:
1.ASH1L在转移性癌症中存在基因扩增及过表达现象
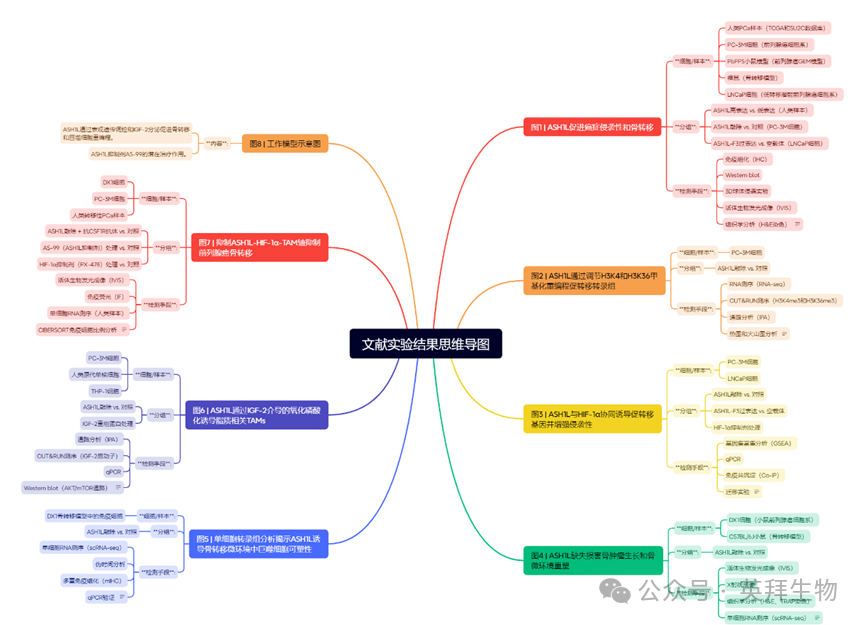
通过对人类癌症的荟萃分析,我们发现ASH1L基因的扩增及增益现象在>40%的转移性前列腺癌(PCa)中出现,其频率显著高于局限性肿瘤(图1a)。高水平的ASH1L mRNA与基因扩增状态相关,且与转移性疾病密切相关(附图1a, b)。此外,ASH1L基因扩增与转移性PCa患者较差的总生存期相关(附图1c),而ASH1L mRNA高表达的人前列腺肿瘤组织呈现富集的癌症转移特征谱(附图1d)。对人PCa肿瘤组织的免疫组化(IHC)分析显示,ASH1L蛋白水平与癌症进展及侵袭性呈正相关(图1b及附图1e)。在人PCa细胞系中,源自PC-3的转移性变异株PC-3M表现出ASH1L表达上调(附图1f)。我们前期研究建立的PB-Cre驱动Pten/Smad4/Trp53联合缺失(PbPPS)转基因小鼠模型26,27,可发展为伴有淋巴结、肺和肝转移的侵袭性前列腺腺癌(附图1g)。研究发现雄性PbPPS小鼠的侵袭性癌细胞及转移瘤中ASH1L高表达(图1c及附图1h)。除PCa外,其他恶性肿瘤中也普遍存在ASH1L基因扩增(附图1i)。具有高转移潜能的乳腺癌和黑色素瘤细胞相较亲本细胞同样呈现ASH1L表达升高(附图1j)。综上,通过人类样本、转基因模型及癌细胞系的系统研究,我们证实组蛋白甲基转移酶ASH1L在转移性PCa及其他恶性肿瘤中存在基因扩增和过表达特征。
我们随后利用CRISPR/Cas9系统敲除PC-3M细胞中的ASH1L基因(图1d),发现ASH1L缺失对细胞增殖或肿瘤生长仅产生适度影响(附图2a, b),但显著降低PC-3M细胞的迁移能力(附图2c)。通过二维和三维侵袭实验证实,ASH1L缺失能明显抑制PCa细胞穿透细胞外基质的侵袭能力——这是转移进程中的关键步骤(图1e及附图2d)。在另一转移性PCa细胞系DU145中也观察到类似现象(附图2e-g)。
为评估ASH1L缺失对骨转移的影响(80%晚期PCa患者会发生骨转移并导致高致残率28),我们向裸鼠心内注射表达红色荧光蛋白(RFP)和荧光素酶的对照组及ASH1L缺失型PC-3M细胞。生物发光成像显示,ASH1L缺失可抑制癌细胞向胫骨、股骨或下颌骨的转移(图1f)。值得注意的是,ASH1L缺失显著延长小鼠的总生存期和无转移生存期(图1g及附图2h)。离体荧光成像显示对照组PC-3M细胞在小鼠骨骼的转移率为67%(12只中8只),而ASH1L敲除组未检测到骨转移(附图2i)。PC-3M细胞建立的骨转移模型通常会在骨微环境中形成溶骨性肿瘤29,组织学分析表明ASH1L缺失能抑制PC-3M细胞在骨骼的定植、减少破骨细胞数量并减轻溶骨性病变(图1h及附图2j)。这些体内外实验证实ASH1L对PCa侵袭及骨转移具有决定性作用。
为鉴于ASH1L基因在转移性疾病中存在扩增和过表达现象,我们进一步探究了ASH1L过表达对PCa细胞迁移、侵袭及转移的影响。与其他高分子量组蛋白甲基转移酶类似30,在哺乳动物细胞中外源表达全长ASH1L蛋白存在技术挑战31,32。为此,我们构建了三个分别编码人ASH1L蛋白1-882(F1)、883-1890(F2)和1891-2969(F3)氨基酸片段的质粒(附图3a, b)。值得注意的是,F3片段包含ASH1L蛋白所有功能域(附图3a),并能逆转ASH1L缺失型PC-3M细胞中H3K36me3和H3K4me3的缺失(附图3c)。此外,在二维和三维培养体系中回补ASH1L-F3片段可部分恢复ASH1L缺失型PC-3M细胞的迁移和侵袭能力(附图3d, e),表明F3片段对维持ASH1L的组蛋白甲基转移酶活性具有必要性和充分性。
我们将ASH1L-F3导入低转移潜能、低表达ASH1L的LNCaP细胞系,证实其能显著增强H3K36和H3K4位点的组蛋白甲基化活性(图1i)。虽然ASH1L-F3过表达不影响细胞生长,但可显著促进细胞迁移和侵袭,而F1或F2片段对细胞迁移几乎无影响(附图3f-i)。为研究ASH1L过表达的体内效应,我们将携带空载或ASH1L-F3的LNCaP细胞注射至裸鼠胫骨,每周进行生物发光成像。如图1j所示,ASH1L-F3过表达显著促进PCa在骨骼中的转移灶生长。ASH1L-F3组小鼠总生存期较对照组明显缩短(图1k)。离体荧光及组织学分析进一步证实,ASH1L-F3过表达不仅促进PCa骨转移灶生长,还伴随新骨形成(附图3j-l)。
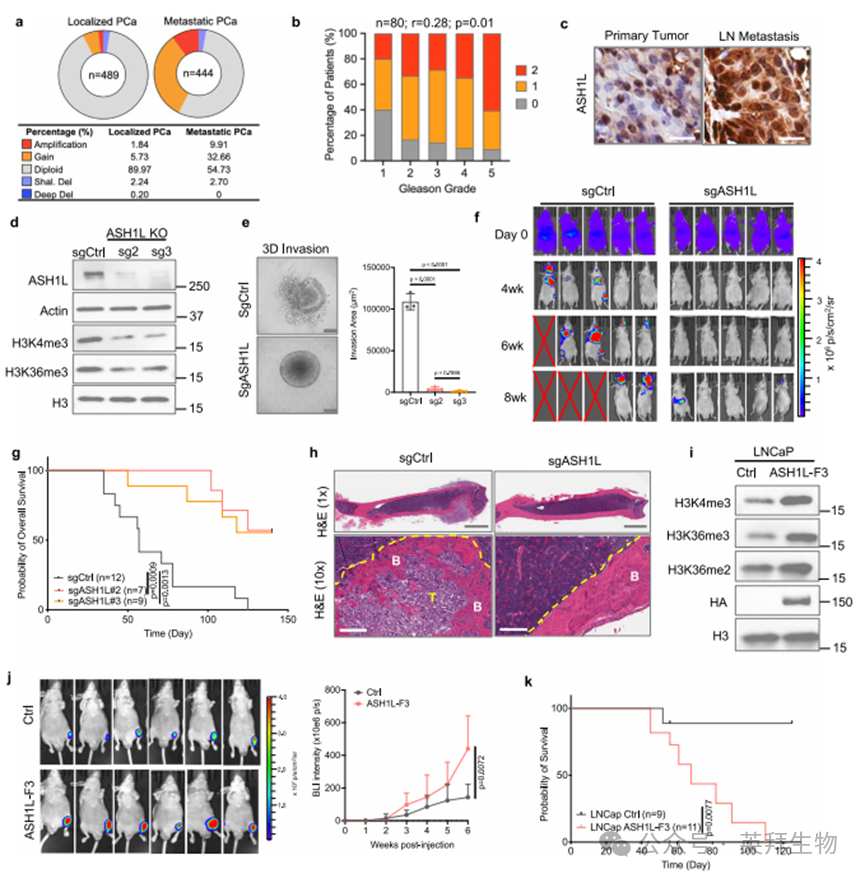
图1.ASH1L在转移性癌症中存在基因扩增及过表达现象
2.ASH1L通过H3K4和H3K36甲基化诱导促转移转录组
为探究ASH1L促转移机制,我们通过RNA测序(RNA-seq)对比分析了对照组与ASH1L缺失型PC-3M细胞的转录组。差异基因表达分析显示,ASH1L缺失导致1378个基因下调及307个基因上调(错误发现率FDR≤0.05,且两个靶向ASH1L的sgRNA均显示倍数变化FC≥2)(图2a-c、附图4a,b及补充数据1),进一步证实ASH1L在转移性细胞中的转录激活作用。Ingenuity通路分析(IPA)表明,这些差异表达基因(DEGs)与EMT(上皮-间质转化)、细胞外基质重塑、整合素信号、血管生成通路及HIF-1α信号等转移相关通路显著相关(图2b)。
已有研究表明,ASH1L通过催化H3K36me2/3和维持H3K4me3参与表观遗传重编程[16-18,20],这些修饰与转录激活密切相关[33-35]。在转移性PC-3M细胞中,我们发现ASH1L缺失导致H3K36和H3K4甲基化水平整体降低(图1d),而过表达ASH1L则增加这些组蛋白修饰标记(图1i)。为深入解析ASH1L对组蛋白甲基化景观的影响,我们采用核酸酶靶向切割与释放测序技术(CUT&RUN-seq)[36],检测了ASH1L缺失前后PC-3M细胞中H3K4me3和H3K36me3的基因组分布。
根据ASH1L缺失引起的组蛋白修饰变化,我们将所有蛋白编码基因分为五个集群(图2d,e及附图4c,d):集群1(C1,占全部基因的3%)显示两种组蛋白修饰信号均降低;集群2(C2,5.3%)仅H3K4me3减少;集群3(C3,3.8%)仅H3K36me3减少;集群4(C4,2%)包含H3K4和/或H3K36甲基化增加的基因;两种修饰均无变化的基因归入集群5(C5,86%)。与预期一致,H3K4me3在全基因组范围内主要富集于基因启动子区(图2d,e及附图4c,d)。ASH1L缺失后,1622个基因(C1+C2,8.3%)的H3K4me3信号强度显著降低(FDR≤0.05;FC≥1.5)(图2d,e及补充数据2)。而H3K36me3信号则分布于基因体区,从转录起始位点(TSS)开始向转录终止位点(TES)逐渐增强(图2d,e及附图4c,d)。ASH1L缺失使1330个基因(C1+C3,6.8%)基因体区的H3K36me3信号显著减弱(FDR≤0.05;FC≥1.5)(图2d,e及补充数据3)。
通过维恩图分析发现,下调的差异表达基因(DEGs)与H3K4me3或H3K36me3信号减弱显著相关。具体而言,46%和35%的下调DEGs(FDR≤0.05;FC≥1.5)分别呈现H3K4me3和H3K36me3信号降低(重叠p值<1e-300,Fisher精确检验)(图2f),提示这些基因(n=1180)很可能是ASH1L在转移性癌细胞中的直接靶标(补充数据4)。热图和箱线图分析显示,集群1-3中H3K4me3或H3K36me3的减少与mRNA水平下调强烈相关,其中集群1的基因表达下降最为显著(图2d,g及附图4e)。值得注意的是,我们发现多个转移相关基因是ASH1L的直接靶标(补充数据4中高亮显示)。ASH1L缺失后,多数靶标基因表现为两种修饰信号同时减弱(如PLAU),但部分基因仅显示H3K4me3(如MMP14)或H3K36me3信号(如VEGFC)的急剧下降(图2h)。需特别指出的是,这三种组蛋白甲基化模式均与促转移基因的转录激活密切相关(图2h)。
这些转录组与表观遗传学研究证实,ASH1L通过重编程H3K4和H3K36位点的组蛋白甲基化,调控侵袭性癌细胞中促转移基因的转录激活。值得关注的是,虽然在白血病中ASH1L通过激活HOX家族基因促进白血病发生[20-23],但在转移性前列腺癌中,ASH1L缺失既不影响HOX基因簇的表达,也不改变其H3K4me3/H3K36me3修饰水平(图2h),表明ASH1L对靶基因的调控具有高度选择性和环境特异性。
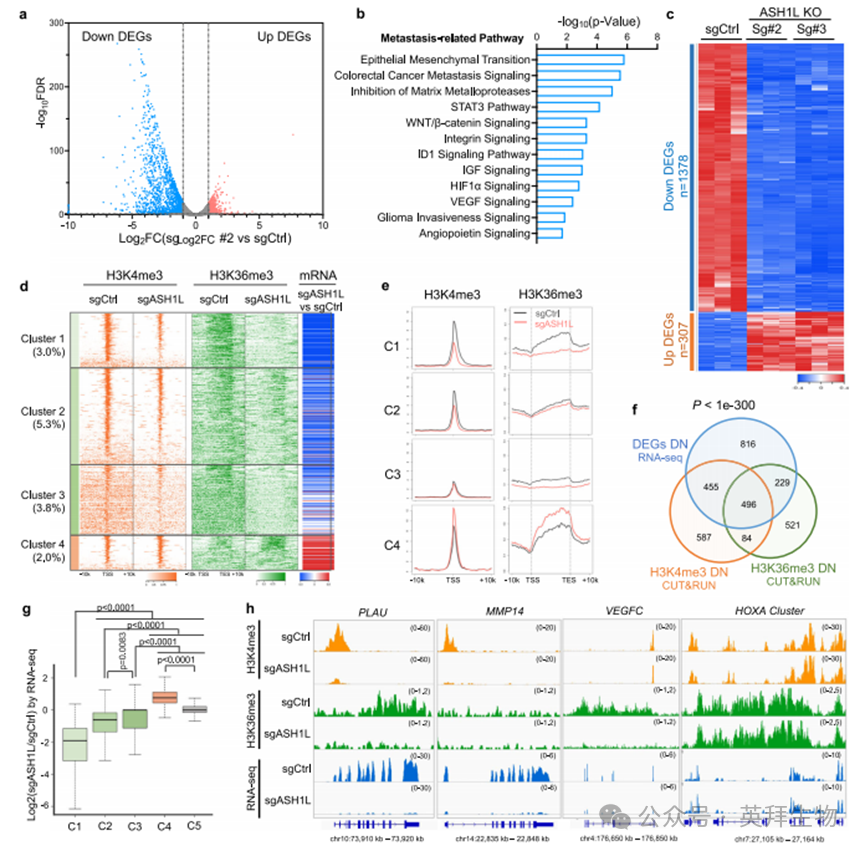
图2.ASH1L通过H3K4和H3K36甲基化诱导促转移转录组
3.ASH1L与HIF-1α协同诱导促转移基因表达并增强侵袭能力
作为缺氧信号通路的核心调控因子,转录因子HIFs能够激活介导血管生成、细胞运动、EMT、细胞外基质降解、侵袭和转移的相关基因表达[38]。HIFs是由氧敏感亚基(HIF-1α、HIF-2α或HIF-3α)与组成型表达亚基HIF-1β构成的异源二聚体。通路分析显示ASH1L与HIF-1α信号通路存在关联(图2b)。基因集富集分析(GSEA)进一步证实,ASH1L缺失会导致缺氧通路及HIF-1α靶基因显著下调(图3a-d),尤其是参与癌细胞侵袭(Snail、TGFB和MET)、细胞外基质重塑(MMPs和PLAU)以及血管生成(VEGFs)的基因。CUT&RUN分析验证了ASH1L缺失后HIF-1α靶基因上H3K4me3和/或H3K36me3修饰标记的显著减少(图3d),表明ASH1L通过调控组蛋白甲基化参与HIF-1α转录程序。
通过对PC-3细胞已发表的HIF-1α ChIP测序数据集(GSE106305)[39]的分析,我们发现83.2%(1180个中的982个)ASH1L直接靶基因(补充数据4)的启动子区域存在HIF-1α蛋白结合,其中包括与癌症侵袭和转移相关的基因(附图5a及补充数据5)。这些结果表明,HIF-1α是ASH1L调控前列腺癌促转移基因的重要协同因子。
此外,qPCR实验显示ASH1L敲除显著下调PC-3M细胞中转移相关HIF-1α靶基因的mRNA水平,而回补ASH1L-F3片段可完全恢复其表达(图3e和附图5b)。在常氧条件下培养的LNCaP细胞因蛋白降解活跃而HIF-1α水平较低,但骨转移灶中的低氧环境能稳定HIF-1α蛋白(附图5c)。为模拟这种缺氧条件,我们用CoCl2处理对照和ASH1L-F3过表达的LNCaP细胞,发现ASH1L-F3显著诱导促转移基因表达(图3f),这与体外和体内观察到的侵袭表型一致(图1j,k及附图3)。相比之下,F2260A突变体诱导促转移基因转录的活性明显受损(附图5d),表明ASH1L的作用依赖其组蛋白甲基转移酶活性。
重要的是,内源性免疫共沉淀(co-IP)和无细胞蛋白下拉实验证实ASH1L-F3直接与HIF-1α蛋白相互作用(图3g和附图5e),强化了ASH1L与HIF-1α在转移性PCa细胞中形成功能复合体的观点。通过分析已发表的人类原发性和转移性前列腺肿瘤样本的批量RNA-seq及单细胞转录组数据[40,41],我们发现HIF家族成员中HIF-1α在上皮成分中与ASH1L的共表达最为显著(附图5f-h)。与ASH1L低表达组相比,高表达ASH1L的PCa肿瘤上皮成分显示HIF-1α转录组水平升高(图3h和附图5i)。
为阐明HIF-1α在ASH1L驱动的促转移基因转录及侵袭中的作用,我们在表达ASH1L-F3的LNCaP细胞中使用siRNA或小分子化合物抑制HIF-1α。结果显示,与对照细胞相比,ASH1L-F3过表达在缺氧条件下引起促转移基因上调,但该效应可被HIF1A敲除所消除(图3i)。同样,用小分子抑制剂(LW6、2-MeOE2和PX-478)靶向HIF-1α能显著削弱ASH1L-F3过表达驱动的LNCaP细胞迁移(图3j),而HIF-2α抑制剂(PT2399)对细胞迁移影响甚微(附图5j)。
我们的机制研究共同表明,组蛋白甲基转移酶ASH1L通过与转录因子HIF-1α相互作用,激活促转移基因转录并增强转移细胞的侵袭能力。值得注意的是,ASH1L在白血病细胞中也与HIF-1α共定位于基因启动子区(附图5k,l及补充数据5),但仅有32.5%(3482个中的1132个)ASH1L靶基因与HIF-1α共享,且不同于转移性PCa中的靶基因(附图5k,l及补充数据5)。这表明ASH1L/HIF-1α相互作用是基因表达的普适调控机制,但其靶基因可能因细胞环境而异。
图3.ASH1L与HIF-1α协同诱导促转移基因表达并增强侵袭能力
4.在免疫健全环境中解析ASH1L的功能
既往研究表明,HIF信号通路在调控原发性和转移性肿瘤的基质与免疫组分中起关键作用[38,42,43]。鉴于ASH1L能促进骨转移并诱导HIF-1α转录组,我们提出假说:除了重编程侵袭性癌细胞的促转移基因外,ASH1L还可能通过细胞外作用重塑骨微环境。基于前期从PbPPS转移性前列腺癌转基因小鼠模型(附图1g)中建立的同源PCa细胞系DX1[44,45],我们通过胫骨内注射DX1细胞至C57BL/6J雄性小鼠构建了同种异体移植模型(图4a),模拟PCa骨转移灶生长。
接种2-3周后,DX1细胞在骨组织中形成兼具溶骨/成骨特征的转移灶(图4b,c及附图6a),重现了PCa患者的骨转移特征。组织病理学分析显示,转移灶中皮质骨显著吸收并伴随大量编织骨新生,同时破骨细胞与成骨细胞明显富集(图4c-g及附图6a)。
为探究ASH1L在免疫健全环境中对转移灶生长及骨微环境的影响,我们通过sgRNA-CRISPR-Cas9系统敲除DX1细胞中的ASH1L基因(附图6b),并将表达荧光素酶的对照组与ASH1L缺失型细胞注射至C57BL/6J雄性小鼠单侧胫骨(图4a)。活体成像(IVIS)及离体GFP荧光成像显示,癌细胞中ASH1L的基因敲除显著抑制骨内肿瘤生长(图4b及附图6c)。X射线成像与组织病理学分析表明,侵袭性癌细胞中ASH1L的缺失可保护皮质骨免于吸收,并减少新生编织骨形成(图4c-e及附图6d)。ASHL1敲除后,骨转移灶中的破骨细胞与成骨细胞数量均减少(图4c,f,g)。使用shRNA敲低ASH1L也观察到类似效应(附图6e-k)。结合异种移植转移模型的发现,这些同源模型实验结果确立了ASH1L作为骨转移灶生长的关键表观遗传调控因子。
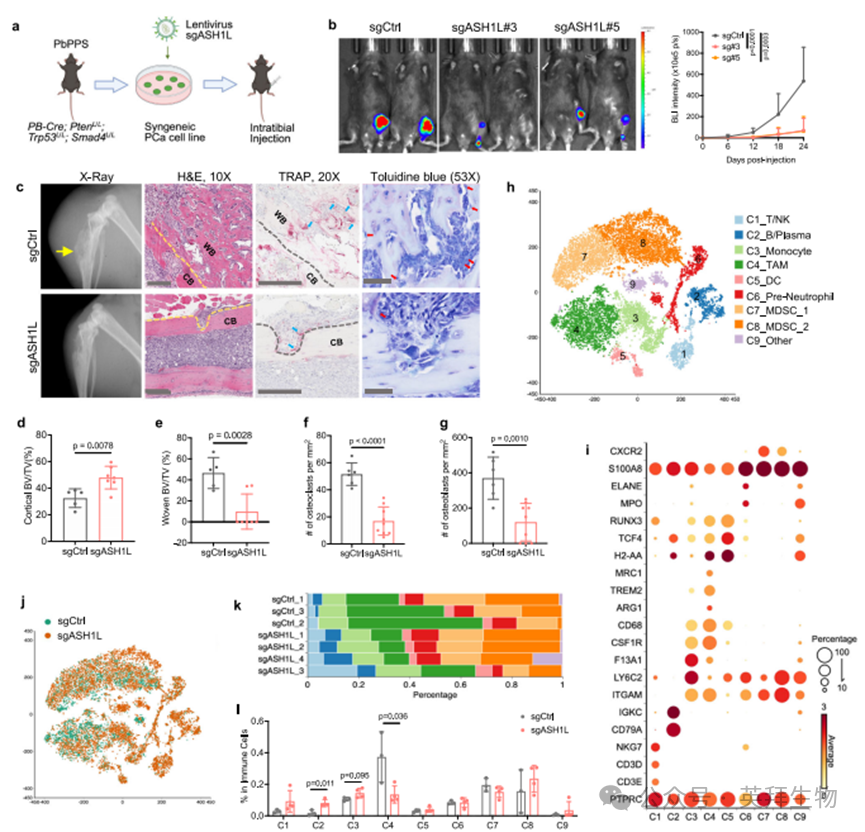
图4在免疫健全环境中解析ASH1L的功能
5.单细胞转录组测序揭示ASH1L调控转移性骨微环境中巨噬细胞可塑性
为解析ASH1L对侵袭性癌细胞及转移性骨微环境的影响,我们对对照组(n=3)和ASH1L缺失组(n=4)同源骨肿瘤进行了单细胞RNA测序(scRNA-seq)。经过标准数据处理与质控流程(方法部分),共获得30,850个单细胞的转录组图谱。聚类分析鉴定出10个主要细胞群(AC1-AC10,每个群含760-6999个细胞;附图6l)。对照组与ASH1L缺失组骨肿瘤细胞分布于所有10个集群,且每个集群均包含7个样本的细胞(附图6m)。通过整合差异基因表达分析和已知细胞谱系标志物(附图6l,n),我们明确了各集群的细胞身份:成纤维细胞群AC3高表达Col1a2,内皮细胞群AC4呈现Pecam1特征,免疫细胞群AC5-AC10高表达各谱系标志物,重现了人类样本的免疫景观。AC1和AC2均显示管腔上皮细胞标志物Krt8高表达,提示为癌细胞群,但仅AC1集群显著高表达HIF-1α转录组特征,且该特征在ASH1L缺失肿瘤中明显减弱(附图6o),强化了ASH1L在调控侵袭性癌细胞HIF-1α转录组中的关键作用。
为可视化ASH1L缺失对骨微环境免疫组分的影响,我们对17,423个免疫细胞进行亚群分析,鉴定出9个主要免疫细胞亚群(图4h-l及补充数据6):T/NK细胞(C1:Cd3ehigh/Cd3dhigh/Nkg7high)、B/浆细胞(C2:Cd79ahigh/Igkchigh)、单核细胞(C3:Itgam+/F13a1high/Ly6c2high)、肿瘤相关巨噬细胞TAM(C4:Itgam+/Csf1rhigh/Cd68high/Mrc1high/Trem2high)、树突状细胞(C5:H2-Aahigh/Tcf4high/Runx3high)、前中性粒细胞(C6:Itgam+/Mpohigh/Elanehigh)、髓源性抑制细胞MDSC_1(C7:Itgam+/S100a8high/Cxcr2high/Ly6c2low)和MDSC_2(C8:Itgam+/S100a8high/Ly6c2high/Cxcr2mid)。值得注意的是,ASH1L缺失增加了B细胞(C2)和单核细胞(C3)的浸润,却显著减少了TAM(C4)的数量(图4j-l),表明ASH1L参与调控转移性骨微环境中的单核髓系细胞。
既往研究显示,肿瘤浸润单核细胞与TAM在微环境中具有异质性,并在癌症进展中发挥不同功能[7,8,10,12,46,47]。为明确其亚型特征与活化状态,并解析ASH1L的影响,我们对对照组和ASH1L缺失组骨肿瘤中的单核细胞(C3)与TAM(C4)进行聚类和差异基因表达分析,最终鉴定出7个特征性亚群(图5a,b及补充数据7):经典单核细胞(MC1)、非经典单核细胞(MC2)、单核/TAM中间态细胞(MC3)、促血管生成TAM(MC4)、脂质相关TAM(MC5)、炎性TAM(MC6)和增殖型TAM(MC7)。对照组与ASH1L缺失组细胞以不同比例分布于所有7个亚群中(图5c,d及附图7a)。
在这些亚群中,MC1和MC2均为单核细胞。与MC2相比,MC1在骨微环境中占主导地位,表达经典单核细胞标志物(如Ly6c2、F13a1、Mgst1和Ccr2);而MC2仅占单核/TAM总数的1-2%,呈现非经典单核细胞特征基因(如Ace、Adgre、Treml4和Cd36)(图5a-d)。亚群MC4-MC7虽均表达巨噬细胞谱系标志物(Csf1r和Cd68),但具有截然不同的转录组特征与活化状态(图5a,b,e,附图7b-d及补充数据7)。MC4亚群高表达促血管生成基因(包括Spp1、Vegfa、Arg1和Mif),故命名为促血管生成TAM;MC5细胞高表达脂质相关TAM标志物(Apoe、C1qc和Trem2)及促肿瘤标志物Mrc1;MC7虽与脂质相关TAM(MC5)共享多数标志物,但呈现高增殖特征(Mki67、Cdk1和Top2a);MC6亚群则富集抗原呈递分子(H2-aa、H2-ab1和Cd40)、干扰素信号通路(IL1rn、Jak2、Stat1、Irf1和Nfkb1)以及T细胞招募/活化相关细胞因子(Cxcl9和Tnf)。值得注意的是,我们还鉴定出MC3亚群,其同时表达巨噬细胞标志物(Csf1r和Cd68)与单核细胞标志物(F13a1和Ly6c2)。轨迹与拟时序分析表明,MC1中的经典单核细胞是其他六个亚群的谱系前体,而MC3细胞处于单核细胞向TAM分化的中间状态(图5f)。
与对照组相比,ASH1L缺失的骨肿瘤中脂质相关TAM(MC5)、增殖型TAM(MC7)和促血管生成TAM(MC4)显著减少,而单核/TAM中间态细胞(MC3)急剧增加,经典单核细胞(MC1)也有轻度增多。差异基因表达分析显示,在转移性癌细胞中敲除ASH1L后,单核细胞与TAM中除TAM标志物外,众多促肿瘤/转移基因和免疫抑制分子也显著下调(图5e,g,附图7b,d及补充数据8)。尽管炎性TAM(MC6)比例变化不大(图5d),但ASH1L缺失后TAM中抗原呈递、干扰素信号和炎症相关基因显著上调(图5e,g,附图7c及补充数据8)。这些结果表明,侵袭性癌细胞中的ASH1L促进骨微环境中TAM向脂质相关、促血管生成和抗炎状态转化。
为直观展示单核细胞与TAM在骨转移灶中的空间分布,我们采用多重免疫组化技术对单核细胞标志物Ly6c、总TAM标志物F4/80及促肿瘤TAM标志物CD206(由Mrc1基因编码)进行共染色。与单细胞转录组分析一致,抑制癌细胞中ASH1L显著减少促肿瘤表型TAM(F4/80+CD206+),同时增加骨微环境中肿瘤浸润单核细胞(Ly6c+)(图5h及附图7e)。反之,癌细胞过表达ASH1L可抑制单核细胞而促进促肿瘤TAM(附图7f)。为验证转移性癌细胞与单核细胞的直接互作,我们从健康人血液中分离单核细胞,与ASH1L敲除或未敲除的PC-3M细胞条件培养基共培养(图5i)。结果显示,ASH1L缺失细胞的条件培养基无法诱导单核细胞表达脂质相关/促血管生成TAM标志物,但促进其高表达炎性TAM标志物(图5i)。在人单核细胞系THP-1中也观察到类似现象(附图7g)。
综合单细胞转录组分析与功能验证,我们揭示侵袭性癌细胞中的ASH1L通过促进单核细胞分化为脂质相关和促血管生成TAM,并将其重编程为促肿瘤和抗炎状态,从而重塑转移性骨微环境。
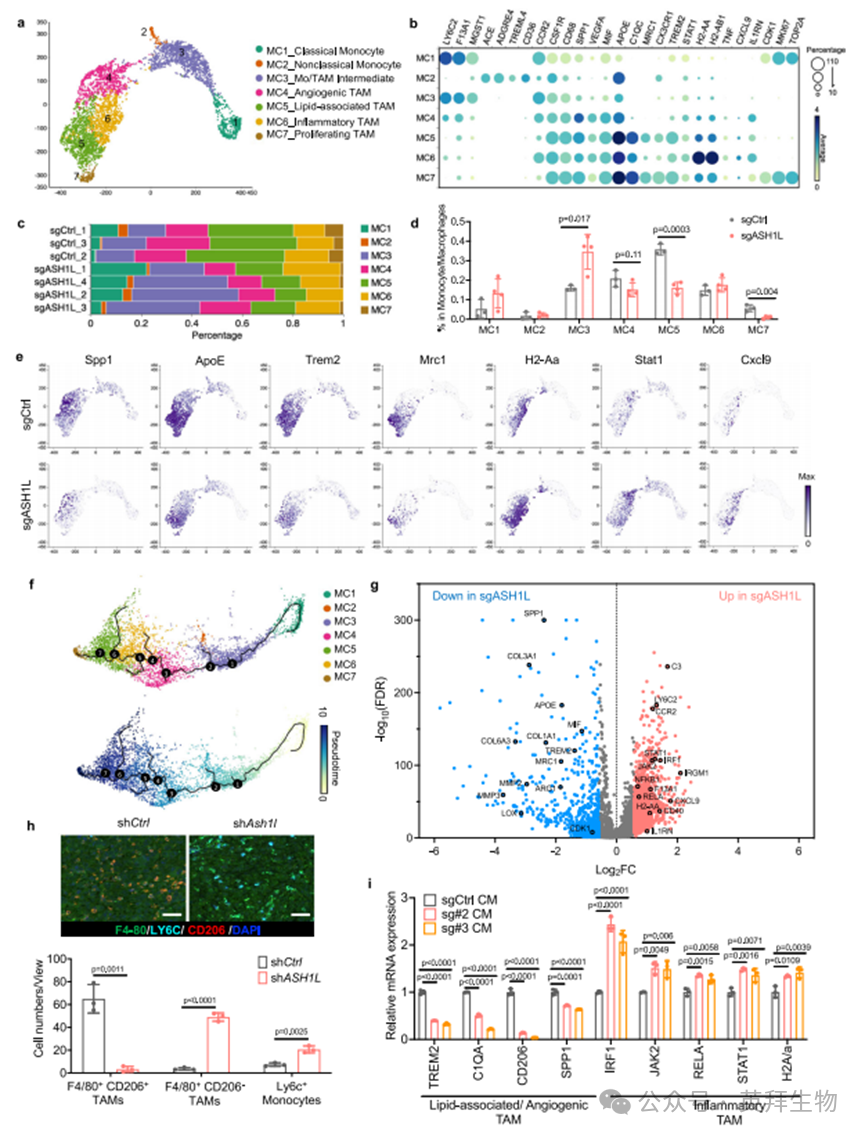
图5 .单细胞转录组测序揭示ASH1L调控转移性骨微环境中巨噬细胞可塑性
6.ASH1L通过IGF-2介导的氧化磷酸化诱导脂质相关TAM形成
为阐明ASH1L调控巨噬细胞分化与可塑性的分子机制,我们对TAM亚群(MC4-MC7)的差异表达基因进行通路分析。与对照组相比,ASH1L缺失骨肿瘤中的TAM显示炎症通路显著激活,包括干扰素信号、TNF信号、IL-1/2信号、cGAS-STING通路以及抗原加工呈递通路(图6a)。同时,ASH1L缺失导致TAM中细胞衰老与焦亡通路相关基因显著上调,而细胞周期与有丝分裂相关基因下调(图6a)。这表明侵袭性癌细胞中的ASH1L通过维持TAM的增殖能力、存活状态及抗炎表型发挥作用。值得注意的是,我们发现转移性癌细胞中ASH1L的缺失会导致氧化磷酸化(OXPHOS)、三羧酸循环(TCA)、脂肪酸β氧化(FAO)和ATP合成功能下降(图6a)。在转移性骨肿瘤的TAM中,OXPHOS特征基因显著富集(图6b),这与既往在原发灶和其他转移部位的研究一致[48,49]。但本研究发现,转移性癌细胞中ASH1L的缺失会显著降低脂质相关TAM中OXPHOS相关基因的表达(图6b)。这些结果表明,ASH1L在重编程转移性骨微环境中巨噬细胞代谢方面起关键作用,促进脂质相关TAM的诱导及其促肿瘤表型形成。
如图5i和附图7g所示,ASH1L缺失的转移性癌细胞条件培养基诱导人单核细胞向脂质相关/促血管生成TAM分化的能力受损,提示分泌因子介导了ASH1L对TAM的调控。为鉴定该因子,我们整合RNA-seq和CUT&RUN-seq数据集(图3及补充数据1-4),筛选出5个TAM相关分泌蛋白候选分子——它们均是ASH1L/HIF-1α的共同靶基因,且在ASH1L缺失的PC-3M细胞中显著下调(图6c)。其中仅胰岛素样生长因子2(IGF-2)与巨噬细胞代谢相关[50]。图6a通路分析也显示,ASH1L缺失骨肿瘤中的TAM呈现IGF下游信号(如AKT和MAPK通路)减弱,提示IGF信号可能参与ASH1L驱动的TAM可塑性与代谢重编程。
IGF家族成员(IGF-1和IGF-2)通过结合受体在细胞增殖、存活、蛋白翻译和代谢中起关键作用[51]。在转移性PC-3M细胞中,IGF-2启动子区富含H3K4me3修饰,但该信号在ASH1L缺失后完全消失(图6d),而IGF-2基因体区未检测到H3K36me3信号,表明ASH1L通过调控H3K4三甲基化调节IGF-2表达。ASH1L缺失还显著降低PC-3M细胞中IGF-2 mRNA水平(图6d,e),反之ASH1L-F3过表达可提升LNCaP细胞IGF-2表达(图6f)。相比之下,PC-3M细胞中IGF-1基因表达极低,且其启动子和基因体均无H3K4me3或H3K36me3修饰(图6d)。ChIP-seq分析显示HIF-1α直接结合PC-3细胞中IGF-2启动子区(附图8a),而siRNA敲低HIF-1α可消除LNCaP细胞中ASH1L-F3对IGF-2的诱导(附图8b),证实IGF-2是转移性细胞中ASH1L/HIF-1α复合体的直接靶标。
单细胞转录组分析显示,转移性细胞中ASH1L缺失导致TAM中OXPHOS复合体I、III、IV和V核心亚基转录水平下降(图6g),这些复合体是催化OXPHOS和ATP合成的关键组分。在ASH1L缺失的PC-3M条件培养基培养的人单核细胞中也验证了这一现象(图6h及附图8c)。在自身免疫疾病中,IGF-2可通过增强OXPHOS使成熟巨噬细胞获得抗炎表型[50]。使用IGF-2重组蛋白处理人单核细胞后,我们同样观察到AKT-GSK3b-mTOR信号激活、OXPHOS基因上调及脂质相关TAM标志物表达增加(附图8d,e)。重要的是,补充IGF-2重组蛋白可基本挽救ASH1L缺失导致的TAM中OXPHOS基因表达及抗炎/促肿瘤表型缺陷(图6h及附图8c)。在转移性PCa患者中,IGF-2表达也与脂质相关TAM标志物及促肿瘤表型标志物呈正相关(附图8f,g)。这些机制研究证实,作为转移性细胞中ASH1L/HIF-1α复合体的直接靶标,IGF-2通过增强OXPHOS介导单核细胞向脂质相关TAM分化,并维持其促肿瘤和抗炎表型。
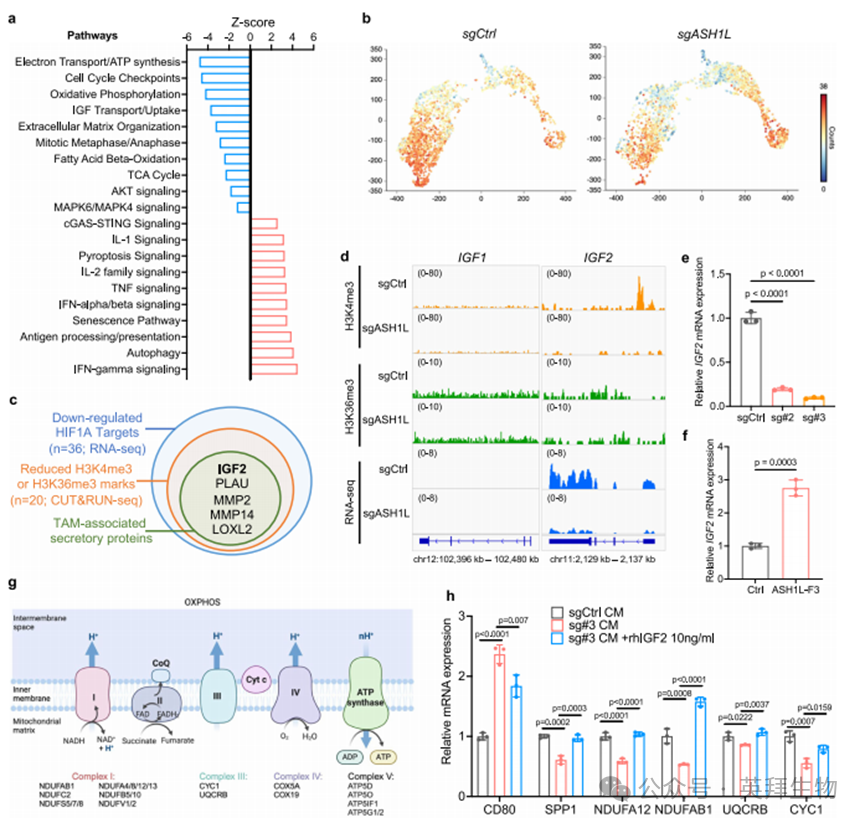
图6. ASH1L通过IGF-2介导的氧化磷酸化诱导脂质相关TAM形成
7.抑制ASH1L-HIF-1α-TAM轴可遏制前列腺癌骨转移
为验证TAM是否在ASH1L驱动的骨转移灶生长中起关键作用,我们开展了体内巨噬细胞清除实验(图7a)。向C57BL/6J雄性小鼠胫骨注射表达荧光素酶的对照或ASH1L敲低型DX1细胞后,每周通过腹腔注射CSF1R单抗或IgG对照抗体(300μg/次)清除巨噬细胞,并进行生物发光成像。与ASH1L缺失表型一致,CSF1R阻断显著抑制前列腺癌骨内生长(图7b,c及附图9a)。值得注意的是,ASH1L抑制仅在巨噬细胞存在时才能削弱骨肿瘤生长,而巨噬细胞清除后该效应消失(图7b,c及附图9a),表明TAM是ASH1L促转移作用的必要条件。免疫荧光染色不仅证实CSF1R阻断后骨肿瘤中总TAM(F4/80+CD206+与F4/80+CD206-)被清除,还验证了ASH1L抑制对骨微环境中促肿瘤TAM(F4/80+CD206+)的削减作用(图7d及附图9b)。
近期研究发现小分子抑制剂AS-99可靶向ASH1L并具有抗白血病活性[52]。在转移性前列腺癌细胞中,我们发现AS-99以剂量依赖性方式降低H3K4和H3K36位点的组蛋白甲基化水平(附图9c),并削弱体外细胞迁移能力(附图9d)。在乳腺癌细胞中也观察到类似效应(附图9e)。随后我们评估了AS-99在前列腺癌骨转移临床前模型中的疗效:将PC-3M细胞注射至裸鼠左心室14天后,分别给予溶剂对照和AS-99治疗(腹腔注射,25 mg/kg,每周5次,持续3周)(图7e)。结果显示AS-99治疗显著抑制骨转移并延长小鼠总生存期(图7e,f)。与对照组相比,AS-99治疗的转移瘤中促肿瘤TAM(F4/80+CD206+)数量减少(图7g及附图9f)。这些临床前研究证实了靶向ASH1L在转移性疾病中的治疗潜力。
鉴于HIF-1α是ASH1L在转移性细胞中的关键协同因子,且靶向HIF-1α的药物已进入临床研究阶段[38,53],我们进一步评估了HIF-1α抑制剂PX-478在骨转移模型中的疗效。通过向C57BL/6J雄性小鼠单侧胫骨注射对照或ASH1L缺失型DX1细胞后,给予溶剂或PX-478治疗(腹腔注射40 mg/kg,每周3次,持续3周)。活体成像(IVIS)与离体荧光成像显示,ASH1L缺失虽能显著抑制骨内肿瘤生长,但该效应可被PX-478治疗所抵消(图7h及附图9g)。值得注意的是,HIF-1α抑制仅在对照组中表现出抗肿瘤效果,而在ASH1L缺失组无效(图7h及附图9g)。多重免疫组化染色进一步证实,HIF-1α抑制剂能显著减少表达ASH1L的肿瘤中促肿瘤TAM(F4/80+CD206+)的浸润(图7i及附图9h)。这些结果揭示HIF-1α在ASH1L驱动的骨转移灶生长及TAM可塑性中起关键作用,提示靶向HIF-1α对转移性PCa患者的治疗潜力。
最后,我们分析了ASH1L与TAM在人类转移性PCa中的相关性。通过对13例转移性PCa患者的24,433个非上皮细胞进行单细胞转录组分析[41],鉴定出代表髓系细胞、淋巴细胞、癌症相关成纤维细胞和内皮细胞的18个亚群(图7j及附图10a,b)。研究发现人类转移性肿瘤中的髓系成分主要为单核细胞和TAM,且所有三个TAM亚群均高表达脂质相关TAM标志物(APOE、C1QA和TREM2),并呈现CD206(MRC1)阳性的促肿瘤表型(图7j及附图10b,c)。根据上皮细胞中ASH1L表达水平,将13例转移瘤分为ASH1L高表达组(n=5)和低表达组(n=8)(附图10d)。与低表达组相比,ASH1L高表达的转移瘤中脂质相关TAM更富集(图7k,l),同时癌细胞中HIF-1α转录组特征也更显著(附图10e)。此外,通过CIBERSORT算法对208例转移性PCa患者的批量RNA-seq数据进行分析显示,存在ASH1L基因增益或扩增的转移瘤含有更高比例的TAM(附图10f)。这些人类样本研究为ASH1L促进转移微环境中TAM(尤其是脂质相关TAM)的作用提供了关键证据。
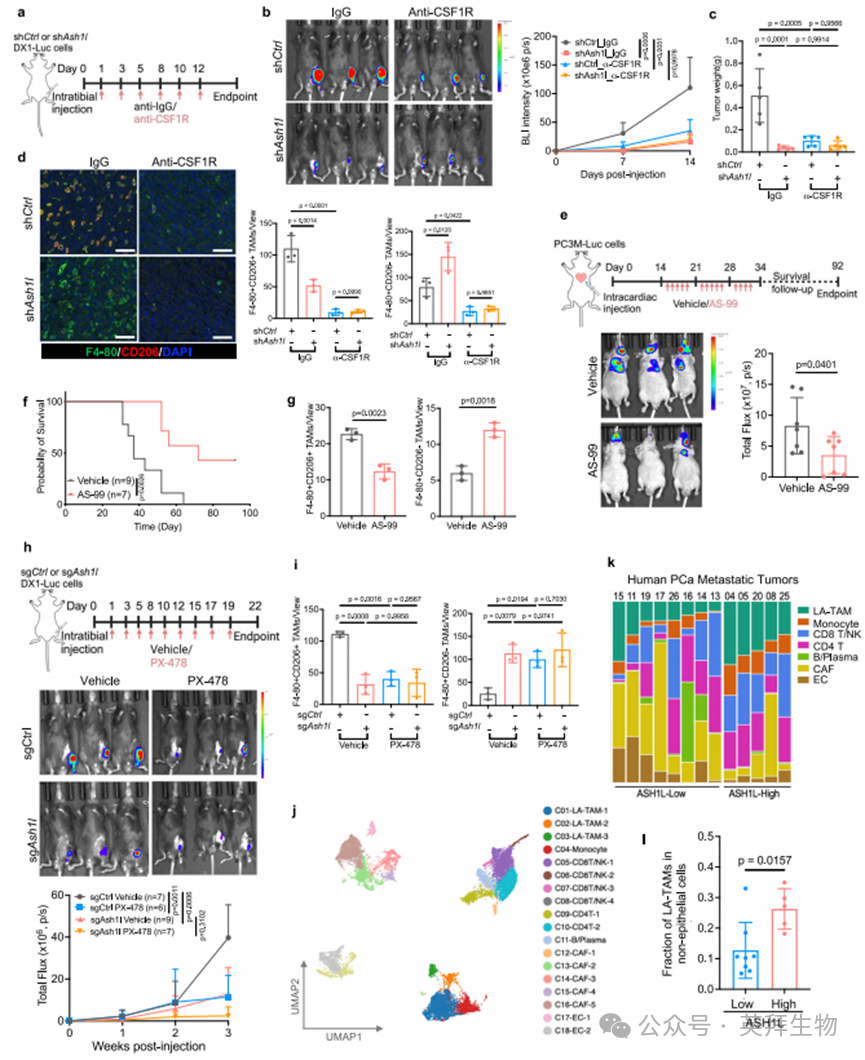
图7 抑制ASH1L-HIF-1α-TAM轴可遏制前列腺癌骨转移
8.分子机制
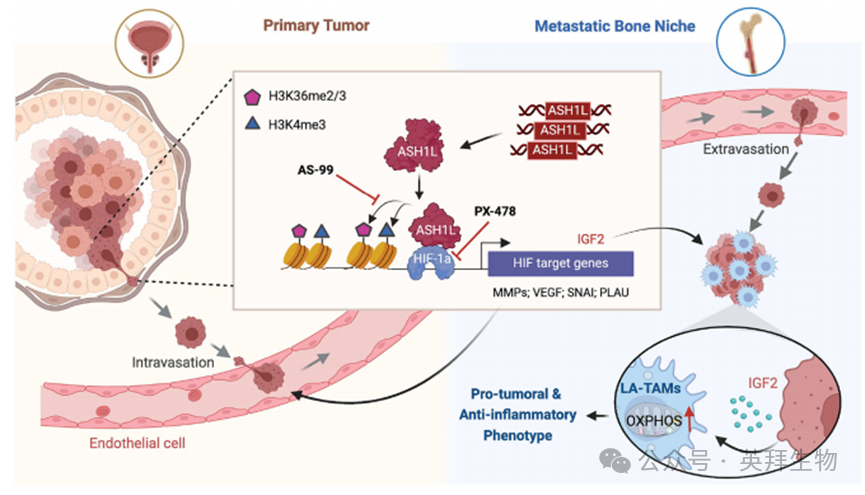
ASH1L在前列腺癌中基因扩增并过表达,通过重编程组蛋白甲基化修饰(如H3K4me3和H3K36me3),与HIF-1α合作,诱导癌细胞中促转移基因表达,促使单核细胞分化为脂质相关巨噬细胞(LA-TAM),增强其促肿瘤表型。此外,ASH1L通过IGF-2介导的氧化磷酸化重编程,促进LA-TAMs的分化和表型变化,从而在骨转移生态位中重塑免疫微环境,促进肿瘤转移和生长。
结论:
组蛋白甲基转移酶ASH1L是骨转移中的关键表观遗传驱动因子。它通过与HIF-1α合作,重编程组蛋白甲基化修饰,诱导癌细胞中促转移基因的表达,并促使单核细胞分化为脂质相关巨噬细胞(LA-TAM),增强其促肿瘤表型。此外,ASH1L通过IGF-2介导的氧化磷酸化重编程,促进LA-TAMs的分化和表型变化,从而在骨转移生态位中重塑免疫微环境,促进肿瘤转移和生长。研究还表明,靶向ASH1L-HIF-1α-巨噬细胞轴的药物治疗能够显著抑制骨转移的发生和进展,为骨转移的治疗提供了新的靶点和潜在的治疗策略。
参考文献:
Meng C, Lin K, Shi W, Teng H, Wan X, DeBruine A, Wang Y, Liang X, Leo J, Chen F, Gu Q, Zhang J, Van V, Maldonado KL, Gan B, Ma L, Lu Y, Zhao D. Histone methyltransferase ASH1L primes metastases and metabolic reprogramming of macrophages in the bone niche. Nat Commun. 2025 May 20;16(1):4681.