






癌旁肺炎症对肺腺癌临床预后的预测作用
摘要
约30%的早期肺腺癌患者在手术切除成功后出现疾病进展。尽管努力绘制基因图谱,但在发现疾病结果的预测生物标志物方面取得的成功有限。在这里,我们对143个肿瘤和匹配的邻近肿瘤、组织学正常的肺组织进行了系统的多组学评估,并对患者进行了长期随访。通过对肿瘤和邻近正常组织的组织学、突变和转录图谱分析,我们发现肿瘤邻近组织中的炎性基因信号是疾病进展的最强临床预测因子。单细胞转录分析表明,进展相关的炎症信号在免疫和非免疫细胞中都有表达,单核细胞中的细胞类型特异性特征进一步改善了预后预测。来自癌症基因组图谱的肿瘤邻近转录数据的额外分析证实了炎性信号与癌症中更差的结果之间的关联。总而言之,我们的研究表明,肿瘤邻近组织的分子图谱可以识别疾病进展的高风险患者。
该研究于2023年11月发表在《Nature communications》,IF:16.6。
技术路线
结果
1、一项匹配的肿瘤-正常肺研究:设计和队列特征
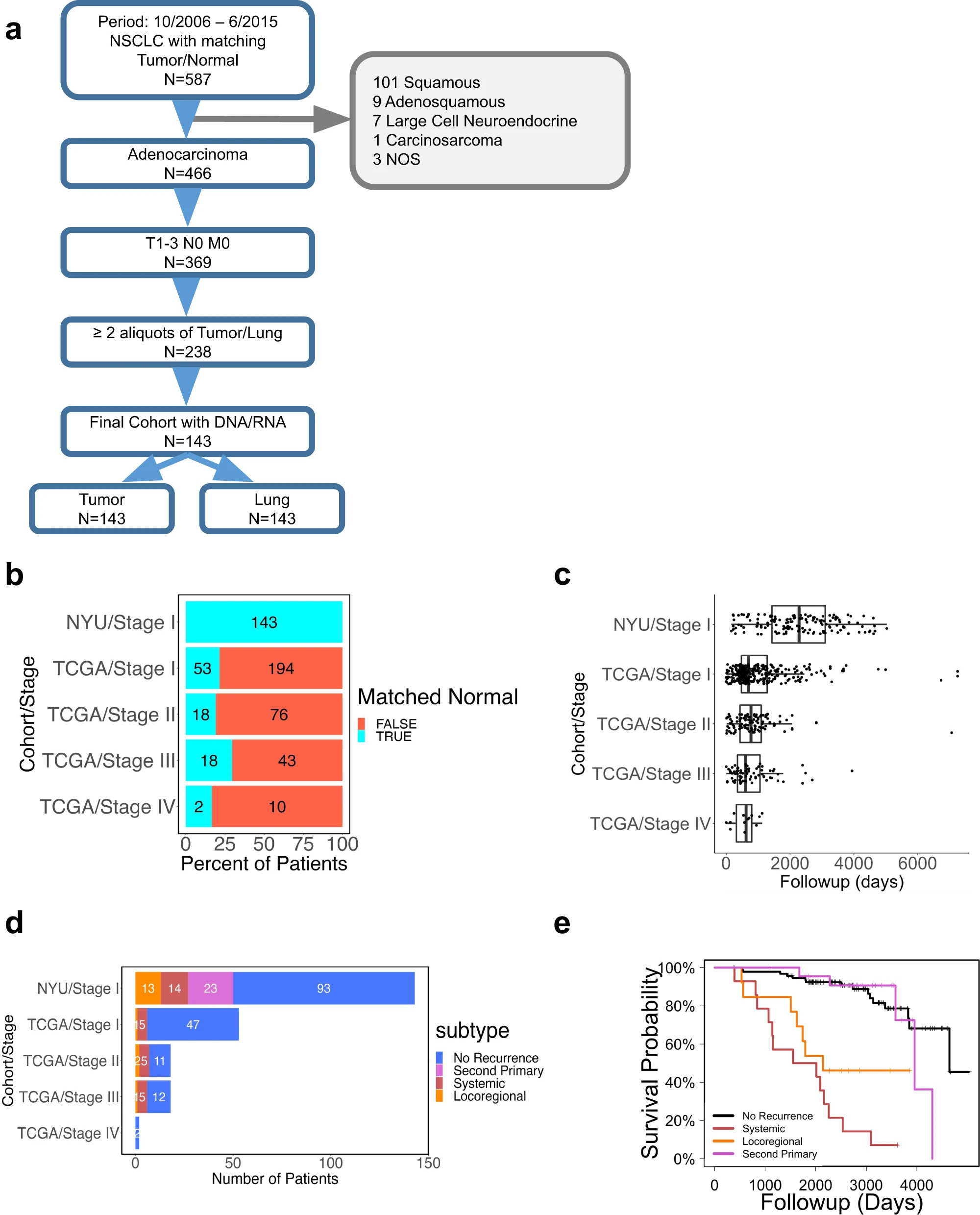
在这项研究中,我们使用了一组治疗初治期Ⅰ的肺腺癌患者,这些患者的肿瘤与肿瘤相邻的正常肺组织标本(在相同的肺叶、肺段或楔形切除内)来自我们的前瞻性收集样本的生物库。纳入研究的患者在手术前从未接受过任何癌症治疗。共有143名患者符合我们的纳入和排除标准(图1A)。据我们所知,这是对匹配的肿瘤-正常早期癌症进行的最大规模的研究,因为TCGA仅限于53名具有匹配的肿瘤-正常样本的I期患者(图1B)。
重要的是,我们的队列有广泛的随访,而TCGA的随访时间相当有限(中位数随访2,284天对701天)(图1C)。长得多的随访使我们能够观察到大量的疾病进展事件,并能够发现无进展生存的分子特征。到目前为止,我们的队列中已经记录了50名(35%)疾病进展的患者。具体地说,我们已经确定了23名患者发生了第二原发于肺的肿瘤,13名患者被诊断为淋巴或肿瘤床局部复发,14名患者存在脑、骨、胸膜、肝脏或肾上腺的系统性转移;相比之下,只有6名患者被记录在TCGA I期队列中有进展的疾病(图1D)。补充图1C-g显示了进展组和非进展组的年龄、吸烟、性别、组织学和国际肺癌研究协会(IASLC)分级的分布。全身或局部复发患者的总体存活率比第二原发肿瘤患者差(图1E)。
2、匹配的肿瘤-正常肺标本的突变和转录图谱
我们首先使用NYU GenomePACT panel 对患者样本进行了DNA测序,该panel涵盖了580个蛋白编码基因的外显子和TERT启动子。对于每个患者,我们使用了来自肿瘤、肿瘤邻近正常(TAN)肺和正常血液的样本。然后,我们对所有286个样本(143个肿瘤和143个肿瘤邻近的正常肺)进行了RNA-seq。15个肿瘤和10个正常肺样本由于文库质量较低,被排除在下游分析之外。最终,123个匹配的肿瘤-正常样本(最初143个匹配样本中的86%)被认为是高质量的RNA-seq样本,并用于下游分析。
3、突变不能很好地预测早期肺腺癌的临床结果
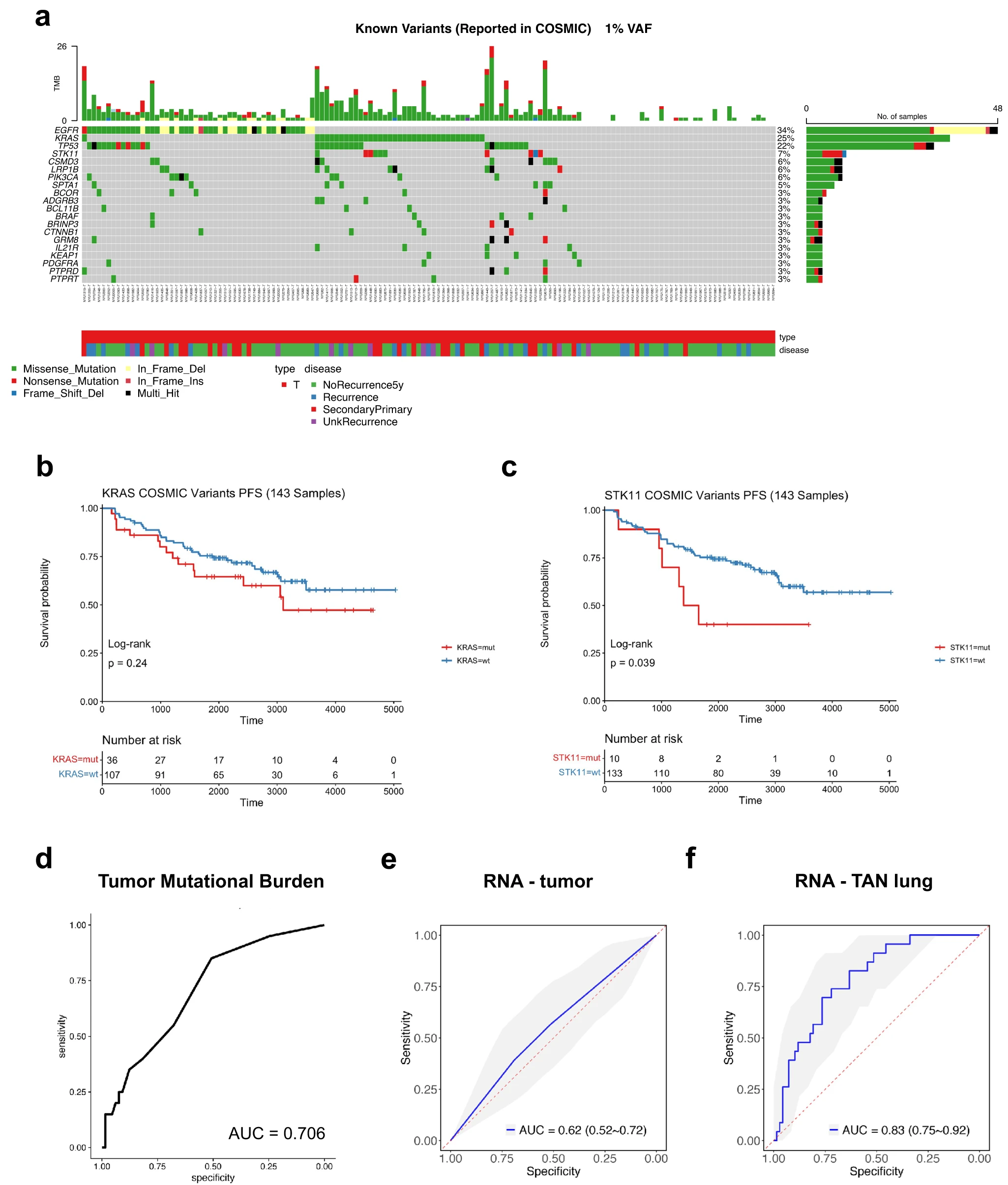
对患者肿瘤DNA测序数据的分析显示,在早期肺腺癌(LUAD)中,常见突变基因的典型分布是:34%的EGFR、25%的KRAS、22%的TP53和7%的STK11(图2A)。然后,我们观察了进展与不进展的患者中可能以不同速度发生突变的基因。我们定义了两组,进展组包括所有疾病进展事件,而不考虑进展类型;无进展组包括所有至少随访5年没有进展的患者。正如预期的那样,按EGFR突变状态对患者进行分层并不会产生PFS的统计学差异,而即使按KRAS或STK11突变状态进行分层也不显著(p值>0.01,图2B,c)。无复发生存率(RFS)也是如此,例外的是与复发显著相关的TP53(p值=0.0053,log-rank检验)。然而,肿瘤突变负荷(TMB)被发现是一个适度的预测5年复发的指标(AUC=0.706)(图2D)。肿瘤邻近正常样本(使用血液作为种系参考)中的突变检测显示,31 个 TAN 样本中变异等位基因频率 (VAF) 截止值为 1% 的突变,其中只有 3 名患者的原发肿瘤中存在突变。此外,只有3名患者有一个VAF高于5%的突变(1名患者有TP53 stop-gain突变,1名患者有非同义PRDM16突变,1名患者有非同义DNMT3A突变),这表明TAN中存在的突变相当有限。综上所述,这些数据表明突变对于 I 期 LUAD 的 PFS 来说是较差的预测因子。
4、肿瘤邻近正常组织中的基因表达具有重要的预后信息
为了确定早期 LUAD 更好的预后标志物,我们随后测试了从批量 RNA-seq 获得的基因表达是否可以提供预后信息并预测 5 年复发。为此,我们构建了一个弹性网络机器学习模型来预测系统性和局部区域的复发,使用嵌套交叉验证来允许自动、无偏的超参数优化,确保从训练到测试集的数据不会泄漏。我们发现肿瘤中的转录组特征不能预测复发(AUC = 0.62,95% 置信区间 = [0.52–0.72])(图 2e),并且不能将患者分为高风险组和低风险组(PFS log-rank检验 p 值 = 0.456)。然而,我们的分析确定,基于 TAN 样本转录组信息的模型显示出优越的性能(AUC = 0.83,95% 置信区间 = [0.75–0.92],见图 2f),并且能够将患者分层为高危人群。和低风险组(PFS 对数秩检验 p 值 = 0.007),显着优于基于肿瘤的模型(Delong 检验,p 值 = 0.0033)。强调在我们的研究中纳入 TAN 样本的重要性,并表明 TAN 肺组织可能导致复发。此外,我们还在 TCGA 的肺癌队列中测试了监督模型。尽管 TCGA 的 TAN 数据有限,但 NYU 模型在 TCGA 肺腺癌 (LUAD) TAN 转录组上具有不错的性能(AUC = 0.75,95% 置信区间 = [0.57, 0.89])。事实上,该模型在应用于 TCGA 肺鳞状细胞癌 (LUSC) 队列的 TAN 转录组时表现同样出色(AUC = 0.74,95% 置信区间 = [0.47, 0.93])。结合这两个队列产生了相似的性能(AUC = 0.75,95% 置信区间 = [0.59, 0.88])。值得注意的是,IASLC 分级在预测进展(AUC = 0.64,95% 置信区间 = [0.56–0.71])或复发(AUC = 0.74,95% 置信区间 = [0.56–0.71])方面表现出显着较低的性能。 [0.65–0.82])。因此,我们的数据表明,基于 TAN 转录组的模型在预测 LUAD 的 PFS、全身性和局部区域复发方面比基于肿瘤的模型具有更强的功效。
5、共表达基因模块分析揭示肿瘤邻近正常肺组织中炎症通路的激活
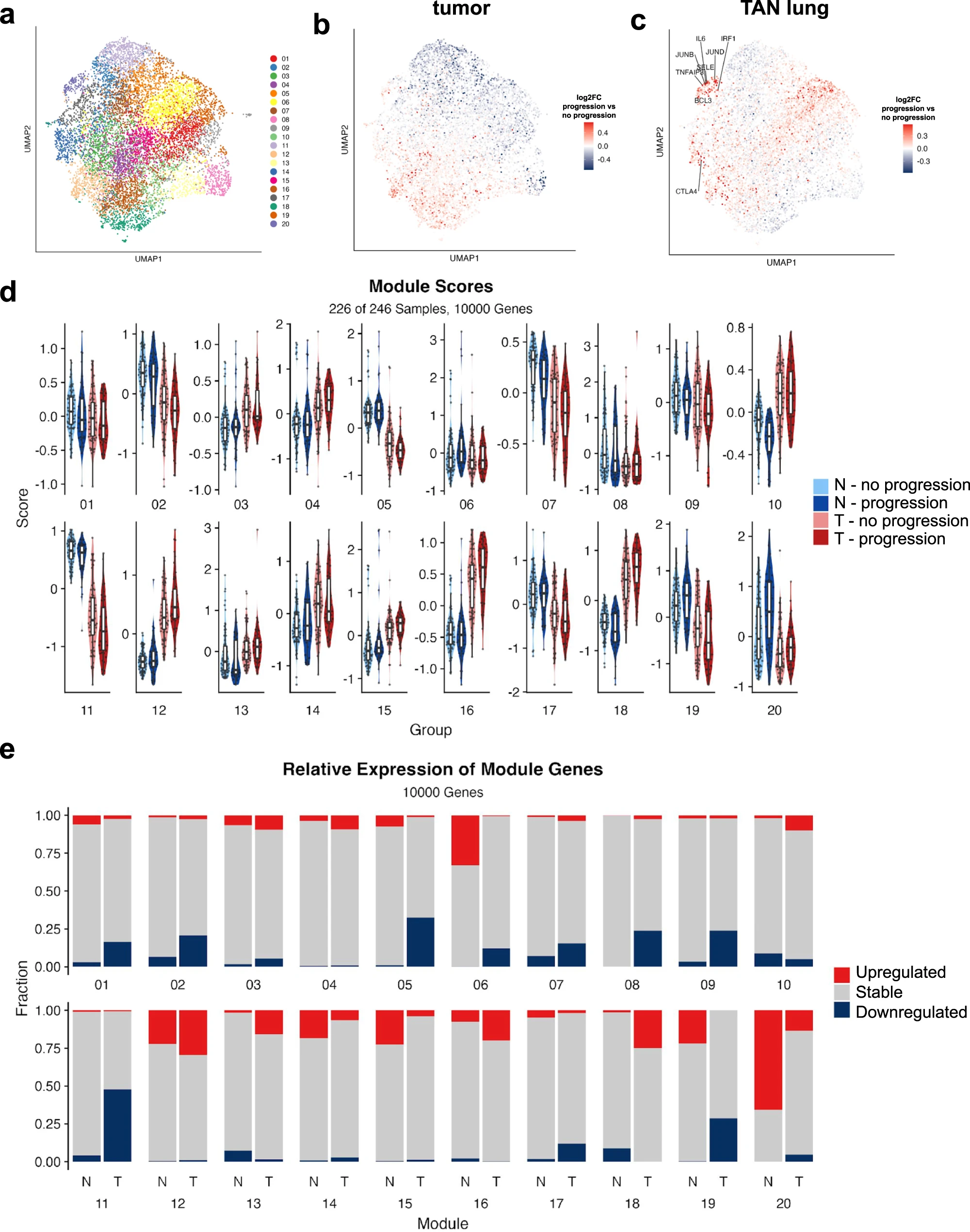
为了进一步了解 TAN 中与肿瘤组织相比具有预后价值的潜在转录程序,我们着手表征 TAN 中特异性激活的转录程序。与其依赖复杂的有监督的机器学习模型(图2e,f),这些模型具有潜在的大量参数和在临床环境中有问题的推广能力,我们决定使用无监督的无偏见方法进一步分析246个匹配的肿瘤正常RNA-seq样本。简而言之,我们选择了前10,000个可变的基因,在样本中缩放它们的表达,并使用Unifold流形近似和投影(UMAP;UMAP上的每个点代表一个基因,详细信息请参阅方法)进行降维。无监督聚类揭示了 20 个基因簇,即共表达基因模块,或者简单地说,模块(图 3a)。然后,我们根据每个基因从 TAN 到肿瘤样本的对数倍数变化对每个基因进行着色,揭示了肿瘤样本中表达较高的基因簇(红色)和正常样本中表达较高的基因簇(蓝色),如图 3b 所示。为了识别与肿瘤相邻正常细胞相比在肿瘤中总体上具有较高表达的模块,反之亦然,我们定义每个模块的分数为模块中基因的平均缩放基因表达(每个患者,每个组织类型)。事实上,我们发现几个模块在正常样本中具有显着更高的平均表达(模块 2、5、6、7、8、9、11、17、19 和 20),而其他模块在肿瘤样本中表达更高(模块 3、4、10、12、13、14、15、16 和 18)(图 3c)。然后,我们根据每个模块与特征、具有明确定义的生物状态或过程的基因集的关联来表征每个模块。被发现与最多特征相关的模块是模块 20(图 3d)。值得注意的是,尽管与肿瘤相比,模块 20 在正常肺组织中的得分更高,但人们发现它显着富集了大量通常与癌症相关的标志,从而证实了肿瘤邻近的正常组织并不完全正常,与之前的研究一致。特别是,炎症信号通路(TNF-α, IL-17, and NFκΒ)、IL-2和IL-6信号、干扰素-γ反应和低氧在模块20基因中高度富集。
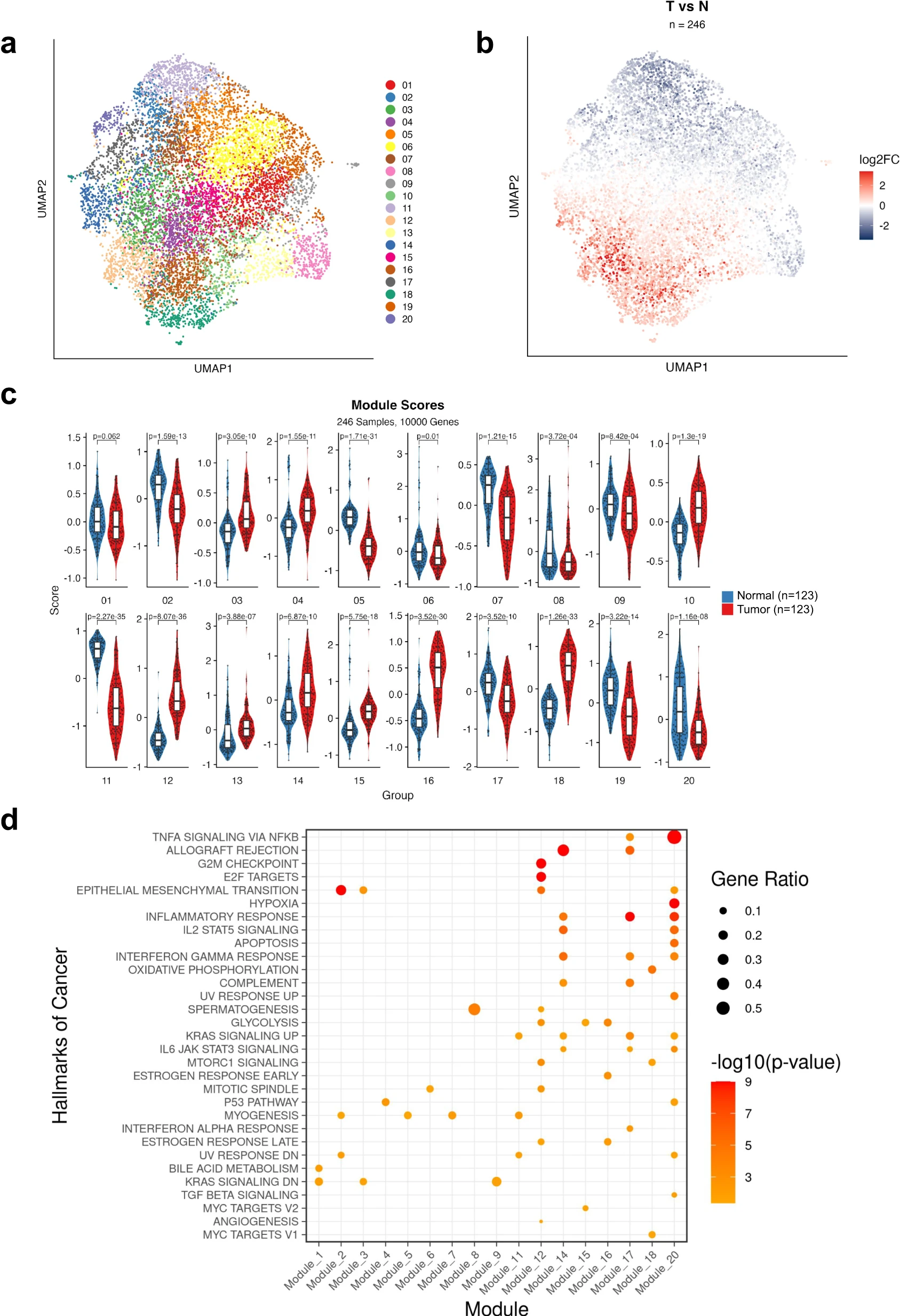
6、肺腺癌在肿瘤及癌旁正常组织中进展的转录转录特征
由于观察到炎症和其他与癌症相关的通路在TAN中被激活,我们假设这些被发现与最多的癌症相关标志相关的通路和相关基因模块的激活(最明显模块20)可能会影响疾病的进展。为了验证这一假设,我们识别了在最终进展的患者组和没有进展的患者组中,在肿瘤或TAN组织中差异表达的基因。更具体地说,来自我们匹配的肿瘤正常队列的患者被分为两组:进展组包括所有有任何类型疾病进展的患者(n=45),而无进展组包括所有在至少5年随访时间内没有进展的患者(n=68)。然后,我们探索了差异表达基因在共表达基因模块中的分布。我们通过分别针对肿瘤(图4B)和TAN样本(图4C)在进展组和无进展组之间表达的对数倍变化来对基因模块UMAP(图4A)中的每个基因进行染色。
对UMAP的可视检查和比较显示,最终进展的患者中上调的基因几乎完全定位在特定的模块中,特别是在TAN样本中。最突出的这类模块是模块20,它在进展期患者的肺脏组织中具有高比例的上调基因。模块聚合表达分析(图4D)证实了这一点,计算了两种组织类型中模块上调和下调基因的百分比(图4E)。显然,模块20高度偏向TAN组织中进展者组中上调的基因,而不是肿瘤中的上调基因。因此,我们的数据表明模块20与TAN和进程有关。
7、一种用于精细化患者分类的多模式关联图
为了进一步确定TAN中已识别的基因模块的特征,我们对模块得分与人口统计学、临床、组织学、遗传学和生存数据进行了全面的关联分析(图5A)。only与不良存活率显著相关的模块是模块20(图5B),在多变量分析(图5C)中,它被发现是临床结果的独立预测因子,对数优势比为0.725(p值=0.002)。有趣的是,作为WHO更新的肺腺癌指南的一部分,IASLC分级在相同的多变量分析中没有发现显著意义。该模型预测复发的敏感性为0.821,特异性为0.491。图5A中的关联图提供了丰富的信息,可用于未来的更大规模的研究,不仅根据人口统计学、临床、组织学和遗传数据的组合将患者分成高度精细的组,而且通过与来自肿瘤和肿瘤邻近正常组织的转录数据相结合,生成关于潜在生物学过程和途径的假设。例如,模块7和10与较年轻的患者相关,与低级别肿瘤广泛相关,缺乏高危组织学模式(实性和融合性颗粒)和较好的预后。模块19和20与老年患者和高级别肿瘤相关,尽管只有模块20被发现与临床结果显著相关。模块8、12和13与胸膜侵犯有关。有趣的是,没有一个模块与突变有关,这支持了我们最初的假设,即肿瘤邻近的正常组织可能是有价值的进展生物标记物的来源,与肿瘤本身的基因构成无关。特别是,模块20的激活发生在患者中,其进展与其肿瘤的驱动程序突变无关。
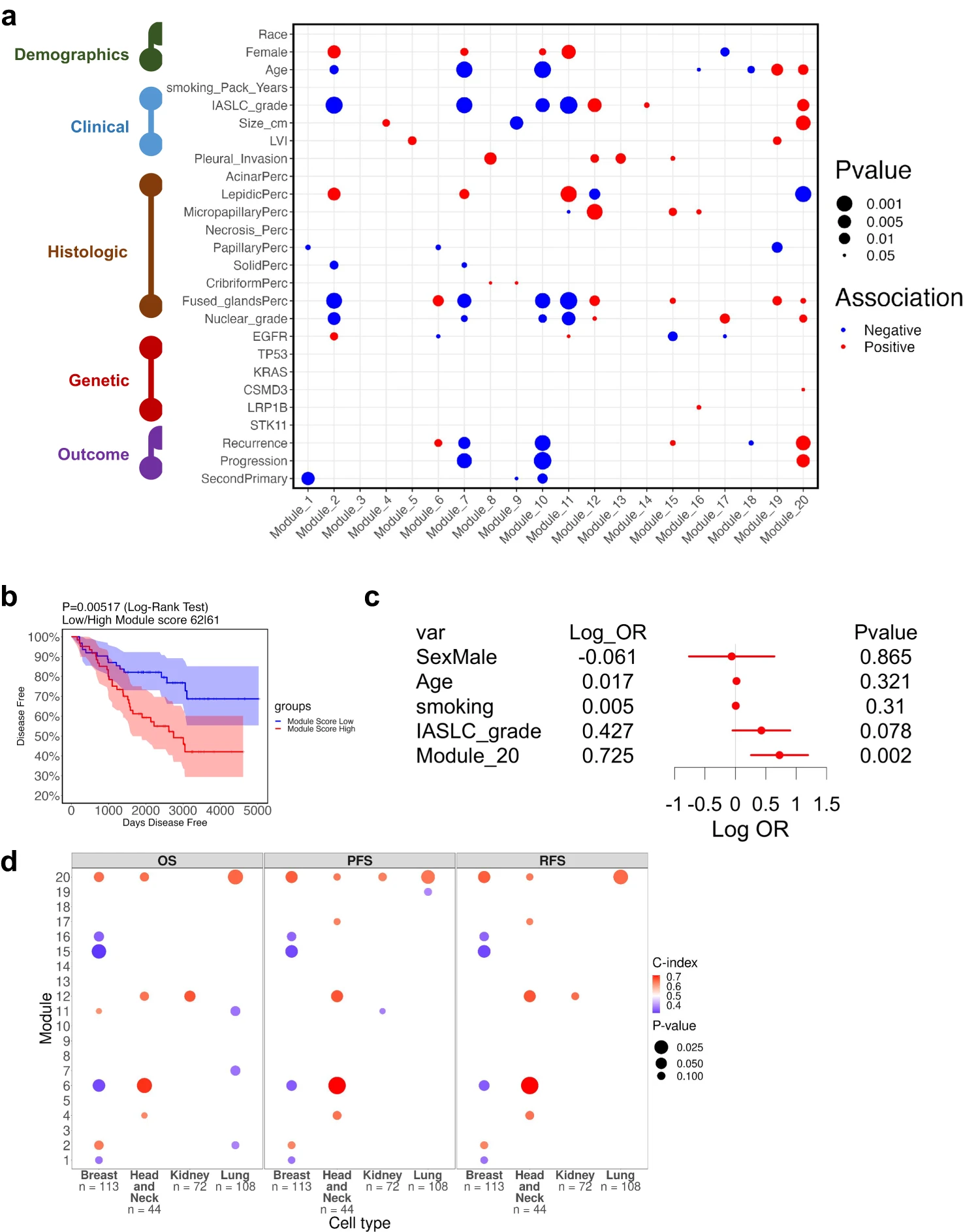
8、其他癌症类型上测试炎症模块20特征
为了进一步测试模块20炎症特征是否可以更广泛地应用于其他癌症类型的TAN组织,我们对从TCGA中的正常组织获得的数据进行了分析。鉴于 TCGA 中使用 RNA-seq 数据的 TAN 样本数量有限,我们只能找到四个原发肿瘤位点,其中至少有 40 个肿瘤相邻正常样本,并且所有阶段至少有两个进展事件:乳腺、肺、肾和头部/颈癌。我们计算了每个模块和每种癌症类型的模块评分与无进展生存率之间的 c-index值(当高模块评分与较差的生存率相关时,c-index值较高)。这项分析的结果如图5D所示,表明在所有四种癌症类型中,模块20是only持续且显著与不良预后相关的模块评分。总而言之,这些发现表明模块 20 在进展中发挥着重要作用。如图3d所示,该模块富含炎症信号传导途径(TNF-α、IL-17和NFκB)和癌症标志(IL-2和IL-6信号传导、干扰素-γ反应和缺氧),尽管它是一个在邻近正常组织中比实际肿瘤更高表达的模块。这一观察表明,最终进展的患者,肺部受损,带有疾病进展的特征,而在邻近的肿瘤中不一定能观察到这些特征。
9、以单细胞分辨率分析肿瘤和肿瘤邻近正常组织
为了确定有助于模块 20 表达的细胞类型,我们利用单核 RNA 测序 (snRNA-seq) 来分析我们的早期肺腺癌匹配的肿瘤正常队列的 TAN 组织。我们对 23 个肿瘤和 23 个匹配的 TAN 样本进行了分析。经过测序后质量控制后,我们剩下 18 个肿瘤样本和 15 个正常 snRNA-seq 样本(112,626 个细胞核)。根据之前对肺腺癌的研究对细胞进行了注释,其中包括正常肺作为对照。重点关注 TAN 样本(51,416 个细胞核),我们鉴定了所有主要细胞类型:上皮细胞、基质细胞、内皮细胞、骨髓细胞、T-NK 细胞、B 淋巴细胞和 MAST 细胞(图 6a) 。不同的细胞谱系被进一步划分为更细粒度的亚群(图6b)。上皮细胞分为四种亚型:肺泡 1 型和 2 型细胞 (AT1/AT2)、球杆细胞和纤毛细胞。基质细胞分为四种亚型:间皮细胞、COL13A1 和 COL14A1 基质成纤维细胞 (FBs) 和周细胞。内皮细胞(ECs)分为三种亚型:淋巴管、茎状和尖状EC。骨髓细胞分为三种亚型:肺泡巨噬细胞、单核细胞和 CD1c DCs。肿瘤样本(61,210 个细胞核)由相同的细胞类型组成,缺乏间皮细胞,并且包含肿瘤细胞,这些肿瘤细胞是根据 inferCNV24 分析计算出的高 CNV 评分进行识别的。
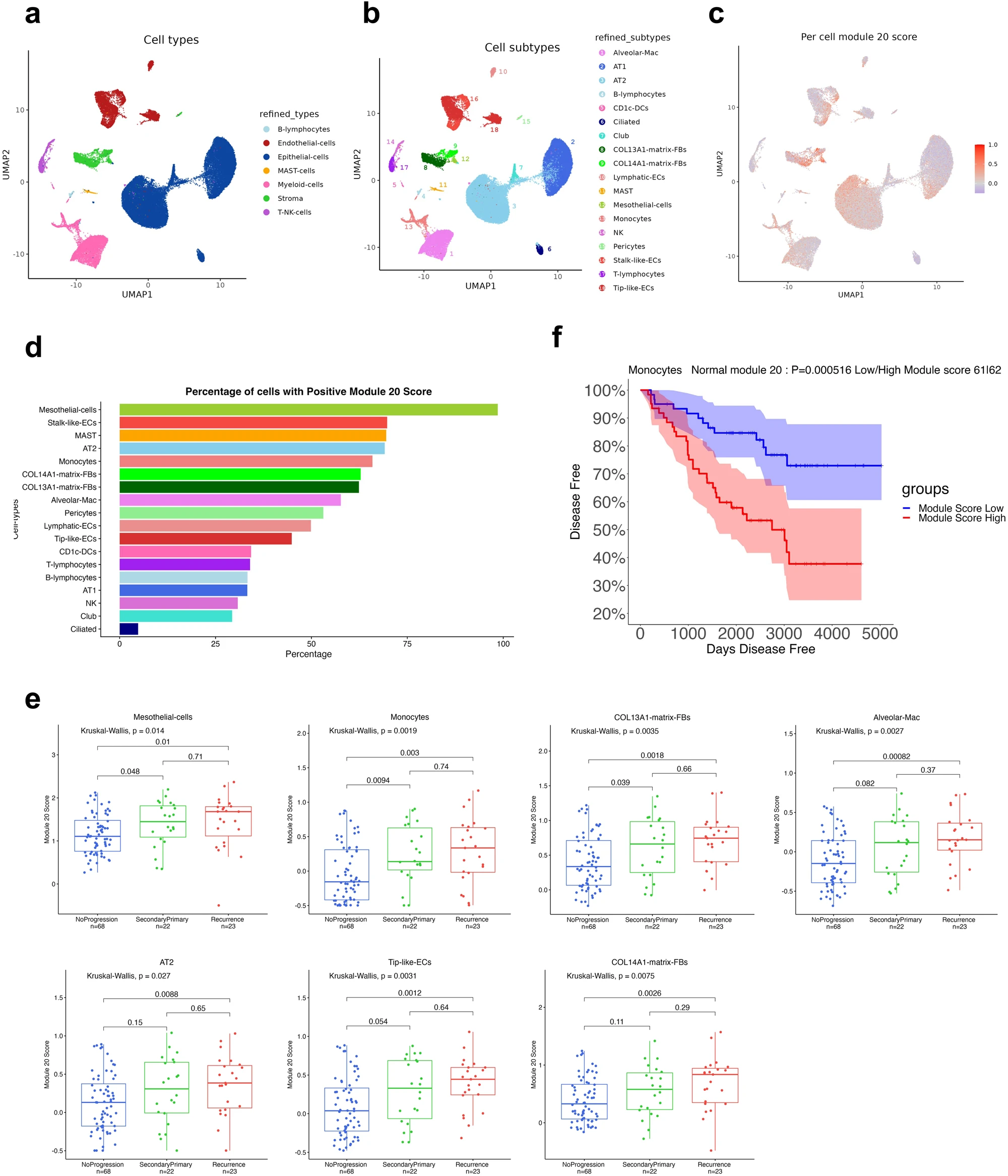
10、模块 20 在进展患者的肿瘤-邻近正常细胞中的多种细胞类型中被激活
为了测试邻近肿瘤的正常肺中哪些细胞类型的模块20中的基因表达升高,我们计算了每个细胞的模块20得分(图6c)。我们观察到,表达模块20基因最高水平的细胞类型是间皮细胞,其次是成纤维细胞、单核细胞、茎状EC、MAST细胞和肺泡巨噬细胞(图6d)。最近,间皮细胞已被证明可以形成抗原呈递的癌症相关成纤维细胞 (apCAF),进而诱导初始 CD4+ T 细胞转变为胰腺癌中的调节性 T 细胞。AT2 细胞(而不是 AT1 细胞)中模块 20 基因特征的激活也很有趣,因为 AT2 细胞已被证明是肺腺癌的起源细胞。
我们测试了哪些细胞类型在进展的患者中上调了模块 20 特征的表达。对于此分析,我们使用了每位患者每种细胞类型的推断基因表达。结果显示,在多种细胞类型中,最终发生第二原发或复发的患者的模块20评分伴随增加(图6e)。值得注意的是,我们对匹配的肿瘤样本进行了相同的分析,并且我们没有观察到任何细胞类型中进展的患者和未进展的患者的肿瘤样本之间的模块20评分有任何显着差异。
我们研究了 TAN 表达模块 20 内特定细胞类型的预后相关性。使用解卷积批量 RNA-seq,我们发现了进展组和无进展组之间模块 20 评分的显着差异。值得注意的是,单核细胞在模块 20 评分中表现出显着差异(p 值 < 0.01)(图 6f),表明它们作为有价值的预后指标的潜力。这些发现强调了考虑TAN表达模块20内的个体细胞类型的重要性,并支持这样的观点:它们可以提供超出整体模块20得分的额外预后能力。
实验方法
DNA测序、RNA测序、机器学习、基因共表达分析、snRNA-seq、Bulk RNA-seq。
参考文献
Dolgalev, I., Zhou, H., Murrell, N. et al. Inflammation in the tumor-adjacent lung as a predictor of clinical outcome in lung adenocarcinoma. Nat Commun 14, 6764 (2023).