






靶向PGLYRP1促进抗肿瘤免疫同时抑制自身免疫性神经炎症
摘要
共抑制和检查点分子抑制肿瘤微环境中T细胞的功能,从而使T细胞失去功能。尽管免疫检查点阻滞是多种人类癌症的成功治疗选择,但严重的自身免疫样不良效应可能会限制其应用。在这里,我们展示了编码肽聚糖识别蛋白1(PGLYRP1)的基因高度与编码共抑制分子的基因共表达,这表明它可能是癌症免疫治疗的有前途的靶点。在小鼠中遗传敲除Pglyrp1导致肿瘤生长减缓,并使CD8+ T细胞呈现出增强的激活/效应器表型,这表明PGLYRP1在CD8+ T细胞中具有抑制功能。令人惊讶的是,遗传敲除Pglyrp1能够保护免受实验性自身免疫脑脊髓炎的发展,这是中枢神经系统自身免疫疾病的模型。PGLYRP1缺陷的髓系细胞在抗原呈递和T细胞激活方面存在缺陷,这表明PGLYRP1在自身免疫期间可能在髓系细胞中起到促炎分子的作用。这些结果突出了PGLYRP1作为免疫治疗的有前途的靶点,当被靶向时,能够引发强大的抗肿瘤免疫应答,同时保护免受一些形式的组织炎症和自身免疫疾病的影响。
该研究于2023年10月发表在《Nature Immunology》,IF:30.5。
技术路线

结果
1、Plyrp1与共抑制基因在T细胞中的共表达
我们实验室以及其他研究组的早期工作表明,在T细胞中,共抑制分子高度共表达在一个基因模块中。因此,我们推断,与共抑制基因在肿瘤中CD8+ T细胞中高度共表达的基因可能编码新的T细胞检查点分子。我们利用了我们先前发表的CD8+肿瘤浸润淋巴细胞(TILs)的单细胞RNA测序(scRNA-seq)数据,这些数据来自B16F10黑色素瘤,以识别共变化的所有基因通过单个细胞的编码已知免疫检查点(Pdcd1、Tigit、Ctla4、Havcr2和Lag3)的基因与干细胞特性相关的基因(Ccr7、Cxcr5、Tcf7和Sell)(图1a)。T细胞中编码多种已知功能分子的基因,包括Cxcr6、Nkg7、Gzmb、Ifng、Prf1、Irf8和Bhlhe40,与检查点基因正相关并与干细胞基因负相关。此外,Pglyrp1一个在T细胞中没有已知功能的基因,也遵循这样的模式(与共抑制分子的Spearman相关系数=0.47,P=1.2×10^-21;与干细胞基因的Spearman相关系数= -0.27,P=1.8×10^-7;P值是用Spearman的渐近t检验计算的)。一致地,Pglyrp1在表达共抑制基因(Pdcd1、Tigit、Ctla4、Havcr2和Lag3)和T细胞功能障碍特征的细胞群中表达,而在表达干细胞样基因(Slamf6、Ccr7、Cxcr5、Tcf7和Sell)的细胞群中不表达(图1b、c)。我们通过实验证实,从MC38-OVA肿瘤中分离的PD-1+TIM-3+ CD8+ T细胞,这些细胞构成了耗竭T细胞,Pglyrp1的表达在体外增加(图1d)。在对来自人类黑色素瘤的免疫细胞进行单细胞RNA测序分析时,我们发现PGLYRP1与其他共抑制分子一起在耗竭CD8+ T细胞中表达最高,尽管其RNA水平较低于编码其他检查点分子的RNA,这表明PGLYRP1与共抑制基因的共表达在耗竭人类T细胞中也是保守的,但可能由于RNA水平较低而没有得到足够的关注。
因此,我们的数据显示,Pglyrp1在小鼠和人类的耗竭CD8+ T细胞中与共抑制基因高度共表达。

2、IL-27对T细胞Plyrp1表达的调节
我们先前已经证明,细胞因子白细胞介素-27(IL-27)诱导了T细胞中的一个共抑制模块。值得注意的是,Pglyrp1是这个由IL-27诱导的共抑制模块的一部分。为了确认IL-27在T细胞中诱导Pglyrp1表达,我们体外培养了有或没有IL-27原生的CD4+和CD8+ T细胞,确认IL-27信号传导后CD4+(约27倍)和CD8+(约2.5倍)T细胞中Pglyrp1表达显著增加(图1e)。先前已经确定转录因子PRDM1和c-MAF是IL-27诱导的共抑制模块的协同调控因子。为了测试它们是否对IL-27介导的T细胞中Pglyrp1表达的诱导起作用,我们体外培养了来自PRDM1/c-MAF缺陷小鼠(Maf–/–Prdm1–/–)的原生的有或没有IL-27的CD4+ T细胞(图1f)。结果显示,IL-27对Pglyrp1表达的诱导在Maf–/– Prdm1–/– T细胞中明显减少,表明IL-27-PRDM1/c-MAF轴在体外诱导T细胞中的Pglyrp1表达。为了检查IL-27和PRDM1/c-MAF是否在T细胞中的Pglyrp1表达中起作用,我们分析了先前发表的CD8+ T细胞的总RNA测序数据,这些细胞是从野生型(WT)和IL-27受体缺陷型(Il27ra–/–)小鼠或Maf–/–Prdm1–/–小鼠的B16F10黑色素瘤肿瘤中分离的(图1g,h)。在这两种情况下,与相应的WT对照相比,我们发现Pglyrp1的表达水平降低。因此,我们的结果表明,IL-27-PRDM1/c-MAF轴在体内外都调控T细胞中Pglyrp1的表达。
3、Plyrp1基因缺陷小鼠中肿瘤生长受到抑制
为了分析PGLYRP1在抗肿瘤免疫中是否起到功能性作用,我们研究了PGLYRP1在不同人类癌症中的表达水平与生存率之间的关联,并发现高表达PGLYRP1的肿瘤患者有明显更差的预后,这表明PGLYRP1在人类癌症中可能作为抗肿瘤免疫的负调控因子。
为了研究PGLYRP1在免疫反应中的作用,我们获得了Pglyrp1–/–小鼠,这些小鼠在稳态下对免疫系统组成和T细胞表型没有改变。为了测试PGLYRP1在小鼠抗肿瘤免疫中的作用,我们植入MC38-OVA结肠癌细胞和B16-OVA黑色素瘤细胞到Pglyrp1–/–小鼠和相匹配的野生型(WT)同窝对照小鼠中(图2)。我们发现植入MC38-OVA细胞的Pglyrp1–/–小鼠的肿瘤大小和肿瘤重量减小(图2a–c),这表明PGLYRP1可能是抗肿瘤免疫的潜在负调控因子。我们还发现Pglyrp1–/–小鼠植入B16-OVA黑色素瘤细胞的肿瘤生长减缓,突显了PGLYRP1缺陷对不同肿瘤类型的肿瘤生长具有保守性作用。
为了了解PGLYRP1可能在肿瘤微环境中对哪种免疫细胞类型发挥作用,我们通过实时定量PCR(qPCR)评估了MC38-OVA肿瘤中分离的不同免疫细胞群体中Pglyrp1的表达。与我们先前的分析一致,Pglyrp1的表达在耗竭CD8+ T细胞中最高。在肿瘤微环境的固有成分中,自然杀伤细胞(NK细胞)和中性粒细胞表达Pglyrp1的水平最高。在MC38-OVA肿瘤中,相对频率方面,Pglyrp1–/–小鼠与野生型小鼠相比,免疫细胞种群没有主要差异,除了Treg细胞在Pglyrp1–/–小鼠的肿瘤中明显减少(图2d)。
因为Pglyrp1与CD8+ T细胞中的共抑制分子共表达,我们接下来集中研究CD8+ T细胞组分。有趣的是,Pglyrp1–/–小鼠的肿瘤和淋巴结中CD8+ T细胞中高频率表达促炎细胞因子,包括干扰素-γ(IFNγ)、肿瘤坏死因子(TNF)和粒细胞-巨噬细胞集落刺激因子(GM-CSF),表明增加了效应功能(图2e)。我们在CD4+ T细胞中没有发现细胞因子表达的差异,除了肿瘤中IFNγ产生的增加。TIM-3和PD-1是共抑制分子,在T细胞激活过程中上调,也是终末耗竭T细胞的标志。我们发现Pglyrp1–/–小鼠的肿瘤、淋巴结和脾脏中CD8+ T细胞中TIM-3和PD-1的表达增加,TIM-3+ PD-1+ CD8+ T细胞的频率也增加(图2f,g)。我们在CD4+ T细胞的TIM-3和PD-1表达没有发现差异。结合我们的功能数据,这些发现表明Pglyrp1–/–小鼠的CD8+ TILs的激活和效应表型增强。
为了全面描述Pglyrp1–/–小鼠中CD8+ T细胞的表型,我们对Pglyrp1–/–小鼠和相匹配的WT同窝对照中的肿瘤浸润CD8+ T细胞进行了bulk RNA测序。我们鉴定了在Pglyrp1–/– 与WT CD8+ T细胞中上调的330个基因和下调的35个基因(FDR<0.15和|log2(fold change)|>1;P值是用edgeR中的似然比检验计算的,经过Benjamini-Hochberg方法(FDR)进行了多重比较调整;图2h)。与我们的流式分析一致,Pglyrp1–/–与WT CD8+ T细胞中表达增加的基因包括参与T细胞激活、效应功能和耗竭基因(Nr4a2、Prf1、Lag3、Irf8、Atf3、Ifng、Havcr2、Tigit和Maf;图2h,i),并富集在T细胞效应和耗竭标志中(图2j,k)。

4、Pglyrp1–/– CD8+ TILs 显示向效应细胞/耗竭细胞转变
为了更好地理解Pglyrp1–/– CD8+ TILs中共抑制分子和效应功能同时增加,我们对来自Pglyrp1–/–小鼠和相匹配的WT对照小鼠的CD45+ CD3+ TILs进行了scRNA测序(图3)。细胞聚类成四个亚群(图3a),我们根据标记物表达将其注释为Treg细胞、CD4+常规T(Tconv)细胞和两个CD8+ T细胞簇。在其中一个CD8+ T细胞簇中,包括与WNT和TCF信号传导以及干细胞和原始样细胞相关的通路富集,表示这个簇为干细胞样细胞。相反,另一个CD8+ T细胞簇富集了T细胞激活、效应功能和耗竭标志,表明一个簇为干细胞样细胞,另一个簇为效应器/耗竭细胞。RNA速度分析表明从干细胞样细胞到效应器/耗竭细胞的轨迹,进一步支持这一模型。与我们早期的分析一致(图1),Pglyrp1的表达在效应器/耗竭和Treg细胞簇中最高(图3b)。
WT小鼠和Pglyrp1–/–小鼠的细胞特征在Treg细胞和CD4+ Tconv细胞簇中混合良好,只有少数差异表达的基因(在Treg细胞和CD4+ Tconv细胞中分别上调8和41个基因,下调4和7个基因;FDR<0.05和|log2(fold change)|≥0.25;P值是用edgeR中的经验Bayes准似然F检验计算的,经过Benjamini-Hochberg方法(FDR)进行了多重比较调整;图3c)。与我们的流式分析一致(图2d),Pglyrp1–/–小鼠的肿瘤中Treg细胞的比例显著减少(图3d)。在WT小鼠和Pglyrp1–/–小鼠的Treg细胞之间差异表达的少数基因中,Itgae(编码CD103)是Pglyrp1–/–小鼠中最高的下调基因(P=1.2×10−5,fold change=2.73;P值是用edgeR中的经验Bayes准似然F检验计算的)。CD103通常高表达于肿瘤浸润的Treg细胞上,并标志着一种特别具有抑制作用的Treg细胞亚群。因此,CD103在Pglrp1-/-Treg细胞上的异常表达可能与其在肿瘤中的低表达有关。
Pglyrp1–/–和WT小鼠之间的CD8+ T细胞特征更加不同,有更多的差异表达基因(239个上调和226个下调;图3c),在主成分(PC)空间中距离更大(最明显在干细胞簇中;图3c),并且有一个明显的全局偏移(图3e)。此外,效应器/耗竭细胞的比例在Pglyrp1–/– TILs中显著增加(图3d),与先前的流式分析一致(图2e–g)。这些数据表明干细胞簇中存在重大表达变化,以及Pglyrp1–/–小鼠中效应器/耗竭细胞比例的增加,表明干细胞样细胞向效应器/耗竭细胞的分化轨迹发生了变化。事实上,Pglyrp1–/–小鼠的干细胞样细胞显示出更高的基因(Stat1、Cd69、Jak2、Tbx21、Irf1和Prf1)和通路(IFN信号传导、白细胞介导的细胞毒作用和耗竭)的表达水平,这些基因和通路参与了T细胞激活和效应T细胞的生成(图3f,g)。这些数据进一步表明了CD8+ TILs中干细胞样细胞向激活/耗竭细胞的转变。
为了检查PGLYRP1在CD8+ T细胞中是否具有细胞内功能,我们生成了有条件的Pglyrp1敲除小鼠(Pglyrp1fl/fl)并将它们与E8iCre小鼠交配,以生成CD8+ T细胞中特异性删除Pglyrp1的小鼠(E8iCrePglyrp1fl/fl)。我们将MC38-OVA肿瘤植入E8iCrePglyrp1fl/fl小鼠和野生型同窝对照动物,并发现E8iCrePglyrp1fl/fl小鼠的肿瘤生长控制更好,并且CD8+ TILs中PD-1和TIGIT表达水平更高的细胞比例增加(图3h,i)。相比之下,特异性在髓系细胞中删除Pglyrp1表达(LysMCrePglyrp1fl/fl小鼠)并没有改变肿瘤生长,这表明Pglyrp1在髓系细胞上的表达对抗肿瘤免疫是不必要的。
总之,我们的数据表明,在CD8+ TILs中删除Pglyrp1导致干细胞样细胞向激活/耗竭细胞的转变,表明PGLYRP1可能是一个调节CD8+ TILs中从干细胞样细胞到效应器的过渡的抑制分子。

5、PGLYRP1缺陷小鼠可免受EAE的侵袭
阻断共抑制分子用于癌症治疗的一个主要障碍是诱导自身免疫样副作用,这些副作用是由于免疫抑制分子在调节免疫反应和恢复体内平衡方面的重要作用而产生的。类似地,在小鼠模型中,阻断和删除T细胞抑制分子,如CTLA-4、PD-1、TIM-3和TIGIT,会导致加重的自身免疫疾病,包括EAE。由于小鼠的肿瘤模型持续时间较短,不允许我们研究自发性自身免疫,因此我们测试了Pglyrp1丧失对自身免疫模型的影响。
为了确定PGLYRP1缺乏是否也会加重自身免疫疾病,我们在Pglyrp1–/–小鼠和匹配的野生型同窝对照小鼠中诱导了EAE。令人惊讶的是,Pglyrp1–/–小鼠在EAE中获得了高度的保护,发病率减少(Pglyrp1–/–:62%;WT:100%),最大临床EAE评分(Pglyrp1–/–:2.3;WT:2.9)和平均临床EAE评分(Pglyrp1–/–:1.6;WT:2.3;图4a–c)。组织学分析进一步显示,Pglyrp1–/–小鼠的中枢神经系统病变和视神经炎减少(图4d,e)。与WT相比,在Pglyrp1–/–小鼠的中枢神经系统中,我们发现造血浸润物(CD45+)以及CD4+ T细胞、CD8+ T细胞、Treg细胞和B细胞浸润物普遍减少,这些细胞类型已知在EAE疾病中发挥重要作用(图4f)。渗透到Pglyrp1–/–小鼠中枢神经系统的CD4+ T细胞表达的炎症因子IL-17A、IFNγ和GM-CSF的水平以及抗炎因子IL-10显著降低(图4g)。渗入的CD8+ T细胞表达的IL-17A和IFNγ水平显著降低。总之,这些数据表明在EAE期间没有PGLYRP1的T细胞启动缺陷。
总之,与其他已知的T细胞抑制分子缺陷的小鼠不同,Pglyrp1–/–小鼠对EAE获得了保护,PGLYRP1似乎对EAE疾病病理起作用。

6、PGLYRP1在髓系细胞中作为促炎分子发挥作用
为了揭示全局Pglyrp1敲除小鼠(Pglyrp1–/–)中减少的EAE表型是由哪种细胞类型负责的,我们使用E8iCrePglyrp1fl/fl小鼠,并将有条件的Pglyrp1敲除小鼠(Pglyrp1fl/fl)与Cd4Cre小鼠交配,以生成CD4+ Tconv细胞、Treg细胞和CD8+ T细胞中Pglyrp1特异性删除的小鼠(Cd4CrePglyrp1fl/fl)。与WT同窝小鼠相比,我们在这两个模型中都没有观察到EAE的发展有明显差异,这表明T细胞中PGLYRP1表达的丧失并不是Pglyrp1–/–小鼠中保护性EAE表型的原因(图5a,b)。
基于在EAE期间Pglyrp1–/–小鼠中T细胞启动缺陷(图4f,g)和在EAE期间调节自反应T细胞启动的髓系细胞的重要功能,我们假设破坏髓系细胞上PGLYRP1的表达可能有助于Pglyrp1–/–小鼠中EAE表型的减少。因此,我们将Pglyrp1fl/fl小鼠与LysMCre小鼠交配,以生成具有髓系细胞特异性Pglyrp1删除的小鼠,包括单核细胞、成熟巨噬细胞、粒细胞和一些树突细胞(DCs;LysMCrePglyrp1fl/fl)。有趣的是,与WT同窝对照小鼠相比,LysMCrePglyrp1fl/fl小鼠对EAE获得了保护(图5c,d),体内的炎症因子IL-6水平较低(图5e)。中枢神经系统内浸润的髓系细胞表现较不活化,如主要组织相容性复合体II类(MHC II类)的表达(图5f)。与减少的髓系细胞活化相一致,dLNs中的CD4+ T细胞似乎启动较少,CD44激活标记物和细胞因子GM-CSF、IL-2和TNF的表达减少(图5g),这表明LysMCrePglyrp1fl/fl小鼠在EAE期间T细胞启动存在缺陷。此外,在中枢神经系统中检测到较低频率的致病性产生IL-17的辅助T(TH17)细胞(IL-17A+ IFNγ+;图5h)。这些数据共同表明,在EAE期间,髓系细胞中的PGLYRP1表达是足够的抗原呈递和T细胞启动所必需的。
为了检验PGLYRP1在髓系细胞中在抗原呈递给CD4+ T细胞中的潜在作用,我们进行了抗原呈递实验。我们将来自LysMCrePglyrp1fl/fl小鼠或WT同窝的脾细胞与具有特异于髓鞘胶质细胞糖蛋白(MOG)的T细胞抗原受体(TCR)的2D2 TCR转基因小鼠的初级CD4+ T细胞在培养基中进行共培养,其中包含MOG肽或不包含MOG肽。在具有LysMCrePglyrp1fl/fl脾细胞的培养孔中,2D2细胞的增殖率较低,并且较低水平的促炎细胞因子IL-1α、IL-6、MCP-1和TNF分泌到培养基中,表明PGLYRP1在髓系细胞中在抗原呈递和CD4+ T细胞启动中起作用。
为了检验PGLYRP1在髓系细胞中是否在EAE期间的T细胞启动中发挥作用,我们在诱导EAE的背景下在LysMCrePglyrp1fl/fl小鼠和WT同窝对照小鼠中进行了一次召回实验。在免疫后的第10天,我们收集脾脏并将脾细胞与或不与MOG肽一同培养。在具有LysMCrePglyrp1fl/fl脾细胞的培养孔中,培养基中检测到较低浓度的促炎细胞因子(IFNβ、IL-6、MCP-1和TNF),这表明PGLYRP1在髓系细胞中在EAE期间CD4+ T细胞启动中起作用。
综上所述,这些数据表明,在EAE期间,髓系细胞需要PGLYRP1进行最佳的抗原呈递和CD4+ T细胞的启动。

7、Plyrp1-/-单核细胞和中性粒细胞的表达变化
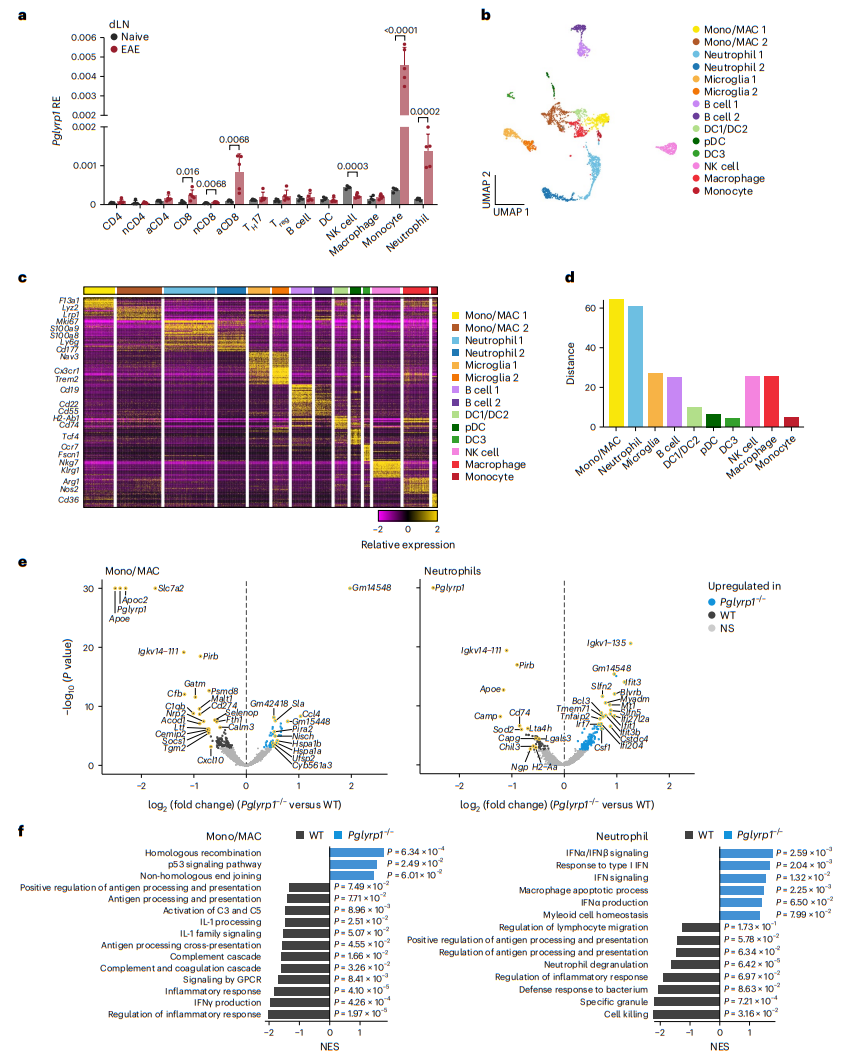
为了揭示在EAE期间PGLYRP1缺陷影响哪些髓系细胞群,我们首先分析了在EAE发作初期的淋巴结内的免疫群体中Pglyrp1的表达情况(图6a)。我们检测到单核细胞和中性粒细胞中表达最高。这种高Pglyrp1表达是由EAE诱导的,因为来自天然小鼠的单核细胞和中性粒细胞显示出较低的Pglyrp1表达。
为了检验PGLYRP1缺陷如何改变髓系细胞组分,我们对来自Pglyrp1–/–小鼠和WT对照小鼠的EAE初发时的CD45+中枢神经系统浸润细胞进行了单细胞RNA测序,并分析了B细胞和髓系细胞(CD3–细胞)。3698个主要是髓系细胞的细胞谱被分为14个簇(图6b),具有明显的表达特征(图6c)。我们使用细胞类型标记和差异基因签名将这14个亚群进行了注释,包括单核细胞/巨噬细胞、中性粒细胞、小胶质细胞、B细胞、DC1s/DC2s、浆细胞样DC、DC3s、NK细胞、巨噬细胞和单核细胞(图6c)。
在每个簇中,Pglyrp1–/–和WT细胞之间的差异(主成分空间中的距离)最大(图6d)。在Pglyrp1–/–和WT小鼠之间的不同基因表达分析中,单核细胞/巨噬细胞(44个基因上调和119个基因下调)和中性粒细胞(163个基因上调和54个基因下调)之间的差异最大(FDR < 0.05且|log2(fold change)| > 0.25;FDR是通过与单核细胞/巨噬细胞相同的方法计算的;图6e)。在Pglyrp1–/–小鼠中,单核细胞/巨噬细胞中减少的基因包括互补系统成员(Cfb和C1qb)和促炎趋化因子CXCL10(Cxcl10)的编码基因。Pglyrp1–/–中性粒细胞中Cd74的表达,该基因编码HLA-DR抗原相关不变链(CD74),较低。有趣的是,Pglyrp1–/–单核细胞/巨噬细胞和中性粒细胞中有九个基因下调,包括Apoe和Lgals3。特别是,在Pglyrp1–/–单核细胞/巨噬细胞(变化排名第1)和中性粒细胞(变化排名第3)中,Apoe显著下调。Apoe编码载脂蛋白E,最近已经显示它在髓系细胞抗原呈递和T细胞启动中具有关键作用,与我们在EAE期间的Pglyrp1–/–小鼠中发现的抗原呈递和T细胞启动的缺陷一致(图4g和图5g、i)。Lgals3编码加凝集素-3,调节单核细胞/巨噬细胞的迁移。在Pglyrp1–/–单核细胞/巨噬细胞和中性粒细胞中,多个与髓系细胞激活有关的基因上调,包括Jun和Lilra6,它们都参与髓系细胞激活。Pglyrp1–/–中性粒细胞中上调的多个干扰素刺激基因(Ifit3、Ifit3b、Ifit1、Ifi204、Ifi27l2a、Oasl2和Irf7)表明了IFN信号的增加。WT单核细胞/巨噬细胞中不同上调的基因富集了互补级联、炎症反应和抗原呈递通路,而Pglyrp1–/–单核细胞/巨噬细胞上调的基因富集了DNA修复和细胞分裂通路(图6f)。WT中性粒细胞富集了涉及杀伤细胞、中性粒细胞颗粒释放和抗原呈递的通路,而Pglyrp1–/–中性粒细胞中富集了与干扰素信号有关的通路。综上所述,这些分析表明,在Pglyrp1–/–单核细胞/巨噬细胞和中性粒细胞中减少的促炎通路和激活,以及巨噬细胞的抗原呈递。
PGN与PGLYRP1结合是其通过TREM-1激活髓系细胞的能力所必需的。为了测试PGLYRP1本身是否也能激活髓系细胞,我们在体外用PGN、重组PGLYRP1或PGN + 重组PGLYRP1处理单核细胞,并测量促炎细胞因子TNF分泌到培养基中。我们发现只有经过PGN和PGLYRP1处理的单核细胞显著分泌TNF,这表明与PGN结合的PGLYRP1介导髓系细胞的激活,在没有PGLYRP1的情况下,髓系细胞不能有效地被激活,从而无法在EAE期间进行最佳的抗原呈递。
总之,我们发现单核细胞和中性粒细胞中Pglyrp1的高表达以及在EAE期间Pglyrp1–/–小鼠中这些细胞群体中出现大量的转录变化。这些数据支持这样一个模型,即在单核细胞/巨噬细胞和中性粒细胞中PGLYRP1的表达对于EAE中致病反应的最佳启动是必需的。单核细胞/巨噬细胞和中性粒细胞的抗原呈递所需功能的缺陷进一步导致了Pglyrp1–/–小鼠和LysMCrePglyrp1fl/fl小鼠EAE疾病的减轻。
实验方法
初代CD4+T细胞的分离与分化、主动诱导EAE、CNS 组织学、肿瘤实验、流式细胞术、荧光激活细胞分选、抗原呈递试验、召回实验、基于微珠的免疫分析、qPCR、Bulk RNA-seq、scRNA-seq、细胞类型注释、RNA速度分析、GSEA。
参考文献
Schnell, A., Huang, L., Regan, B.M.L. et al. Targeting PGLYRP1 promotes antitumor immunity while inhibiting autoimmune neuroinflammation. Nat Immunol (2023).