






URI重编程p53野生型肝癌脂质代谢,减轻TKIs诱导的铁死亡!
基于酪氨酸激酶抑制剂(TKIs)的全身治疗晚期肝细胞癌(HCC)的临床获益受到耐药性的限制。在这里,作者发现由非传统的前折叠蛋白RPB5相互作用因子(URI)介导的脂质代谢重编程使HCC对TKIs诱导的铁死亡具有抗性。在机制上,URI直接与TRIM28相互作用,并以依赖TRIM28- MDM2的方式促进p53泛素化和降解。重要的是,p53结合硬脂酰辅酶A去饱和酶1(SCD1)启动子并抑制其转录。URI高表达与SCD1高表达相关,二者协同表达预示HCC预后不良和TKIs耐药。SCD1抑制剂aramchol与氘化索拉非尼衍生物多纳非尼联合在p53-野生型HCC患者来源的类器官和移植肿瘤中显示出良好的抗肿瘤作用。这种联合治疗对于具有野生型p53和高水平URI/SCD1的晚期HCC患者具有潜在的临床益处。本文于2023年10月发表于《Nature Communication》,IF:16.6,Q1。
技术路线

主要实验结果
1、URI促进肝癌细胞对TKIs诱导的铁死亡的抵抗
首先,作者进行了RNA测序(RNA-seq)分析,以确定激活URI依赖性转录组改变。作者发现在shRNA(Ctrl)和shURI HepG2细胞之间共有1676个基因差异表达。使用KEGG途径进行的功能富集分析显示,铁死亡是URI调控的最重要的改变途径之一(图1a)。基因集富集分析(GSEA)显示,与对照组相比,shURI细胞中铁死亡而不是凋亡信号呈阳性富集(图1b)。这些结果提示URI可能在铁死亡中起重要作用。同时,包括EGFR酪氨酸激酶抑制剂耐药、脂解调节、p53信号通路和癌症中的转录失调等途径也受到URI的调节(图1a)。
有许多临床试验评估TKIs治疗晚期HCC。与细胞毒性作用一致,这些TKIs在测试的各种癌细胞系中有效地降低了肿瘤活力(图1c)。值得注意的是,以B-RAF为共同靶点的索拉非尼、瑞格拉非尼和多纳非尼有利于铁中毒样细胞死亡,而其他测试药物对细胞脂质过氧化的影响很小(图1d)。
然后,作者测试了URI是否调节TKIs诱导的细胞毒性。增加索拉非尼浓度处理JHH1和HepG2细胞48小时,观察细胞增殖情况。URI的消耗显著增加了JHH1和HepG2细胞对索拉非尼的敏感性,在JHH1细胞中,IC50(导致50%的活力抑制)从8.324 μM(Ctrl)降低到6.480 μM(shURI),在HepG2细胞中,分别从8.047 μM(Ctrl)降低到4.069 μM(shURI)(图1e)。克隆形成实验证实了URI在索拉非尼耐药中的作用(图1f, g)。有趣的是,在JHH1和HepG2细胞中,URI敲低显著增加了索拉非尼诱导的脂质过氧化(图1h),这表明URI可能抑制TKIs诱导的铁死亡。此外,索拉非尼诱导的URI敲低细胞的细胞毒性作用可以通过抗氧化剂N-乙酰半胱氨酸(NAC)或铁死亡抑制剂铁抑素-1(fer1;脂质过氧化清除剂),而细胞凋亡、坏死或自噬抑制剂作用不大(图1i)。总之,这些结果表明URI与肝癌细胞对TKIs诱导的铁死亡的耐药性有关。
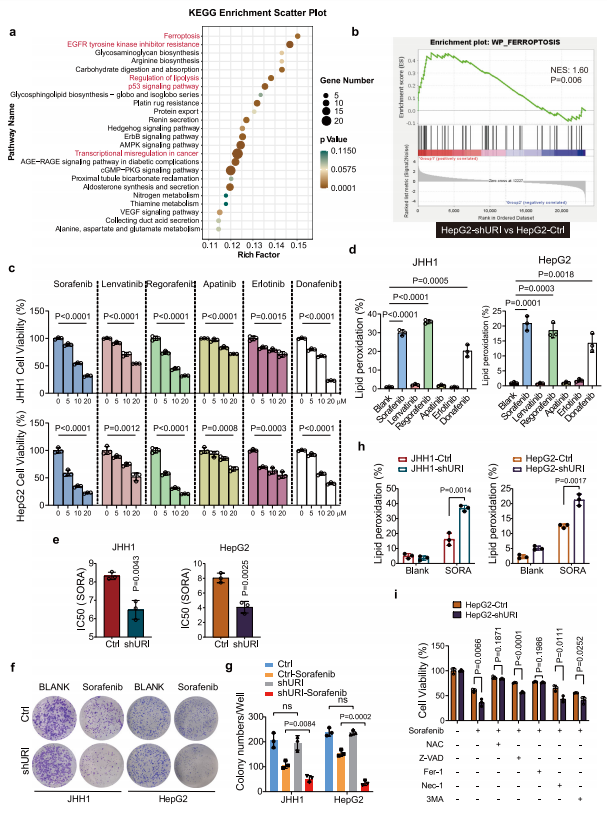
图1 URI耗竭促进TKIs诱导的癌细胞铁死亡
2、URI的异位表达重编程了癌细胞中SCD1相关的脂质代谢
癌细胞比正常细胞需要更高水平的脂质代谢,这可以决定细胞对铁死亡的敏感性。为了检测URI介导的脂质代谢和脂质组学变化,在HepG2- shURI和HepG2- Ctrl细胞中进行了基于质谱的脂质组学分析(图2a)。在URI缺失的HepG2细胞中,主要含有单不饱和脂肪酸链的最丰富的脂类TGs的相对含量比对照组显著降低,磷脂酰肌醇(PI(18:1/18:1))和双甘油酯(DG(16:1/18:1/0:0))的相对含量降低(图2a, b)。相反,磷脂酰甘油(PG(16:0/16:0))的相对含量在HepG2- shURI细胞中增加(图2b)。这些结果表明,URI的消耗改变了肝癌细胞的脂质组成。
作者分析HepG2细胞的脂肪酸含量。细胞内细胞脂质过氧化是铁死亡的一个主要事件,它是由MUFAs和多不饱和脂肪酸(PUFAs)的比例决定的。作者发现,在HepG2- shURI细胞中,SFAs水平比对照组升高,SFAs衍生的代谢产物MUFAs水平,特别是16:1(n-7)和18:1(n-9)MUFAs水平显著降低(图2c)。这些结果强烈表明,URI敲除导致从SFAs到MUFAs的约定受损。值得注意的是,作为SCD1活性替代物的16:1(n-7)/16:0和18:1(n-9)/18:0比值在HepG2-shURI细胞中比对照显著降低(图2d),表明URI表达与SCD1活性/表达之间存在强烈关联。
PL-PUFAs易受ROS影响,其脂质过氧化可引发铁死亡级联反应。相反,MUFAs可以通过促进PUFAs从血浆膜中位移膜磷脂来抑制这一过程。然后,作者分析HepG2-shURI和HepG2-Ctrl细胞之间磷脂的脂质种类(如PC, PE, PI)。稳态下,HepG2-shURI细胞的磷脂中MUFA比对照组有降低的趋势(图2e)。HepG2-shURI细胞中C16:0/C20:4 PL-PUFA含量比对照细胞增加,而HepG2-shURI细胞中PL-MUFA C16:0/C18:1含量降低(图2f)。因此,虽然HepG2-shURI和对照细胞之间PUFAs没有明显变化,但PL-PUFA减少。
从头合成脂肪酸(FAS)涉及几种酶的协调作用(图2g)。接下来,作者测量JHH1和HepG2细胞中FAS通路关键酶的mRNA水平,以及它们的URI耗尽酶(图2h)。与亲本细胞相比,JHH1-shURI和HepG2-shURI细胞中参与饱和脂肪酸合成或脂肪酸延伸的酶,包括SCD1、FASN、FADS2、ACACA和ELOVL6的转录本显著减少(图2h)。总之,这些结果表明URI消耗重编程了肝癌细胞的脂质代谢。
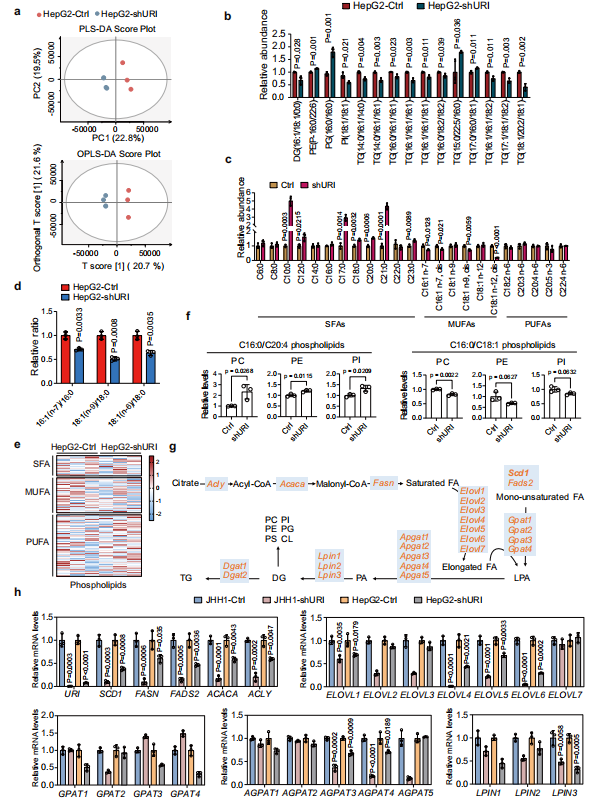
图2 URI耗竭改变了癌细胞的脂质代谢
3、URI通过SCD1促进对TKIs诱导的铁死亡的抗性
脂质代谢重编程与癌症耐药有关。为了探究URI缺失是否会改变脂质代谢相关酶的蛋白水平,作者分析不同细胞系在基础状态下的蛋白水平。虽然在URI敲低的细胞中观察到ACACA、FASN和FADS2的信使RNA转录显著变化(图2h),但它们的蛋白水平不受URI的影响(图3a)。值得注意的是,作者发现shURI细胞中的SCD1蛋白水平低于对照组(图3a),这与SCD1 mRNA水平的降低是一致的(图2h),这表明URI可能通过影响SCD1的转录来调节其活性。
cystine-import-GSH-GPX4机制是调控铁死亡的典型途径。然而,URI缺失在RSL3处理或不处理的JHH1和HepG2细胞中诱导SLC7A11、GCLC、GCLM和GPX4蛋白水平轻度变化(图3a)。此外,在shURI和对照细胞中,ferrostatin-1可以减少索拉非尼或联合治疗诱导的细胞死亡(图3b)。与这些结果一致, SCD1抑制剂与索拉非尼联合在长期克隆实验中显示出对肿瘤细胞增殖的协同抑制。抑制铁死亡可以减轻这种联合治疗诱导的增殖抑制(图3c, d)。进一步的实验表明,索拉非尼和SCD1抑制剂联合使用显著提高了癌细胞的脂质过氧化,加入ferrostatin-1同样减少了联合治疗诱导的脂质过氧化(图3e)。为了证实这些发现,作者将MOCK或Flag-SCD1质粒稳定转染到癌细胞中。作者的研究结果表明,在URI敲除细胞中,Flag-SCD1的稳定表达足以重新抵抗细胞的铁死亡(图3f)。同时,补充外源性SCD1可降低索拉非尼诱导的脂质过氧化(图3g)。总之,作者的研究结果表明,URI通过上调SCD1来促进对TKIs诱导的铁凋亡的抗性。
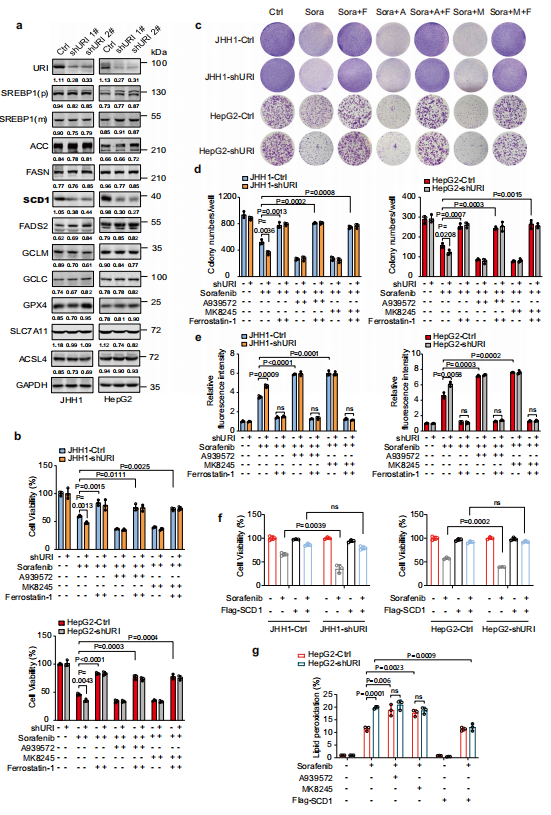
图3 URI以SCD1依赖的方式减轻TKI诱导的铁死亡
4、URI在肝癌细胞中通过野生型p53正向调节SCD1转录
研究表明,SCD1在转录水平上受几种转录因子(TFs)的调控,在翻译后水平上受蛋白酶体的泛素化和随后的降解的调控。用His-URI质粒转染shURI细胞可恢复SCD1蛋白水平(图4a)。为了研究URI如何上调癌细胞中SCD1的表达,作者首先研究了URI对SCD1蛋白稳定性的影响。这些结果表明,URI上调SCD1表达独立于泛素蛋白酶体系统。因此,作者假设URI可能通过上调其转录来促进SCD1的表达。事实上,报告基因分析显示,shURI细胞中的SCD1-luc值显著低于Ctrl细胞(图4b)。
为了进一步确定哪些转录因子介导URI对SCD1的调控,作者将靶向SREBP、ChREBP或LXRα的siRNA转染到癌细胞中,这是报道的调节SCD1的主要TFs。在干扰这些TFs后,URI介导的SCD1-luc强度差异并未消除(图4c)。由于ChREBP和LXRα显示以SREBP依赖的方式增加SCD1的转录,作者在HepG2- Ctrl和HepG2- shURI细胞中使用SREBP抗体进行了染色质免疫沉淀(ChIP)分析,发现SREBP在SCD1启动子区域有多个结合位点(图4d)。然而,SREBP与SCD1启动子的结合不受URI表达的影响,与荧光素酶报告基因实验一致。作者的数据表明这些转录因子不参与URI调节SCD1。
先前的研究表明SCD1基因是p53介导的转录抑制的靶标。与此一致的是,作者的CUT&Tag分析显示p53的结合位点存在于SCD1的启动子区(图4e)。GSEA发现URI缺失后p53转录基因网络显著阳性富集(图4f),RT-PCR进一步证实了这一点(图4g)。为了验证p53介导的SCD1表达的作用,作者用多柔比星(一种著名的p53激活剂)处理JHH1和HepG2细胞(表达野生型p53)。结果显示,Dox处理后SCD1蛋白和SCD1 mRNA表达降低(图4h, i)。接下来,作者将Mycp53、突变体p53- R273H或p53- R249S转染到p53-null细胞(Hep3B)中,发现野生型p53显著下调SCD1蛋白水平。相比之下,DNA接触突变体(p53-R273H)和构象突变体(p53-R249S)都不影响SCD1蛋白水平(图4j)。事实上,SCD1基因的5 '侧翼区域包含一个与p53结合序列一致的位点(图4k)。ChIP分析进一步表明,内源性p53结合在SCD1基因的启动子区域(图4l)。此外,Dox和nutlin-3都可以激活p53,但不能逆转p53-敲低状态下SCD1蛋白水平的升高(图4m)。作者推测URI上调SCD1表达是由p53介导的。URI缺失增强了p53与SCD1启动子区域的结合(图4n)。接下来,作者将p53的siRNA转染到HepG2-Ctrl和HepG2-shURI细胞中,发现p53的敲低消除了shURI介导的SCD1抑制(图4n)。作者的数据显示,URI缺失显著降低了野生型p53细胞(JHH1和HepG2)中SCD1的表达(图3a和图4p)。总之,这些数据表明URI通过抑制野生型p53促进SCD1转录水平。
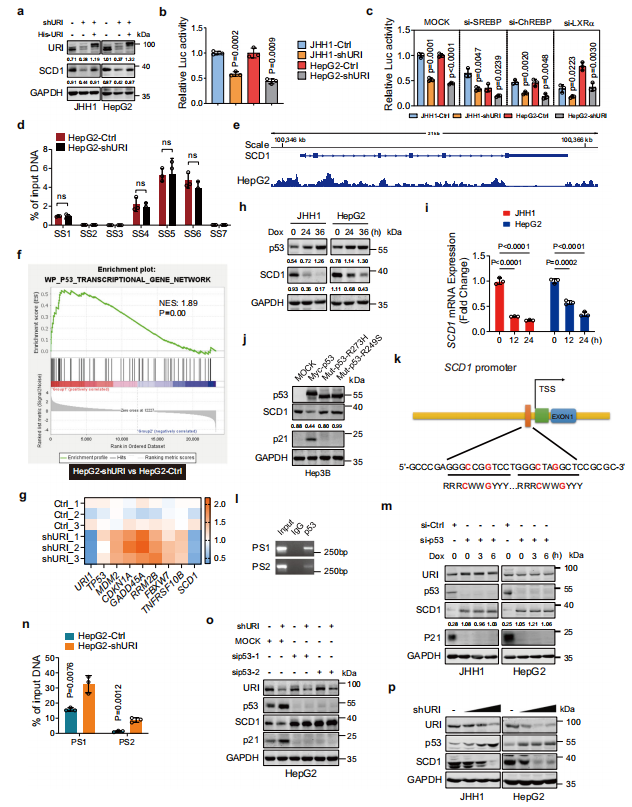
图4 URI通过抑制p53促进SCD1转录
5、URI促进癌细胞中野生型p53的泛素化和降解
接下来,作者将His-URI质粒转染到JHH1和HepG2细胞中,发现这种处理导致p53蛋白显著减少,即使在Dox存在的情况下也是如此(图5a)。为了探究URI介导的p53表达降低的分子机制,作者首先检测了Ctrl和shURI细胞中p53的mRNA水平。作者的数据显示,URI缺失不会影响p53的转录水平(图5b)。应激诱导的p53激活主要通过蛋白稳定发生。作者进一步探讨URI是否调控p53蛋白的稳定性。事实上,与对照细胞相比,使用环己亚胺(CHX)可以延长JHH1-shURI细胞中p53的半衰期(图5c)。URI缺失显著降低了HepG2细胞中野生型p53的泛素化(图5d)。
由于URI不是E3泛素连接酶,作者假设URI可能会招募E3泛素连接酶来介导p53的泛素化。作者进一步进行了液相色谱串联质谱(LC-MS/MS)分析,并确定了潜在的URI结合蛋白,包括TRIM28、USP5、USP7和USP14,这些蛋白被报道可以调节p53的泛素化(图5e)。为了验证LC-MS/MS的结果,作者通过免疫沉淀法测试了候选蛋白与URI的结合。结果显示URI和TRIM28之间存在相互作用(图5f),这与之前的研究结果一致。两种不同的靶向TRIM28的siRNAs增加了p53蛋白水平,特别是在shURI细胞中(图5g)。此外,TRIM28的敲低导致MG132处理下p53的泛素化降低(图5h)。由于TRIM28与MDM2的结合有助于p53蛋白的稳定性,作者探索URI是否会影响TRIM28与MDM2的相互作用。免疫沉淀显示URI的缺失抑制了TRIM28-MDM2的形成(图5i)。相反,URI的过表达促进了MDM2和TRIM28的合作(图5j)。此外,URI缺失降低了TRIM28- MDM2 -p53复合物(图5i, j),表明URI可能募集E3泛素连接酶TRIM28,促进TRIM28- MDM2对p53蛋白泛素化的作用。体外通过GST-pull down实验确定URI是否与TRIM28直接相互作用。纯化后的GST-URI与His-TRIM28之间的相互作用以浓度依赖性的方式显著增加(图5k)。总之,作者的研究结果表明URI与TRIM28相互作用并促进TRIM28- MDM2 -p53复合物的形成,随后泛素化p53。接下来,作者确定URI/TRIM28对p53蛋白水平的调节是否对铁死亡有功能影响。TRIM28的敲低显著促进索拉非尼诱导的细胞死亡和脂质过氧化(图5l-n)。
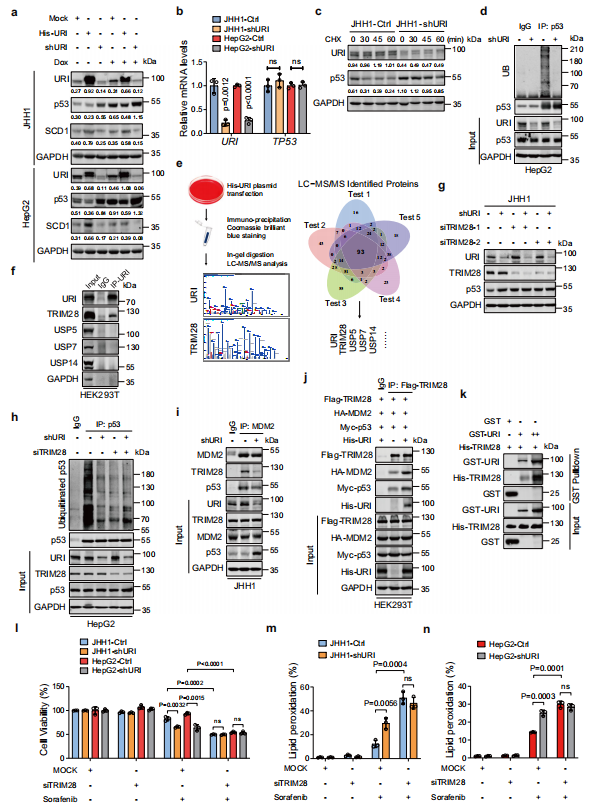
图5 URI通过与TRIM28相互作用促进p53泛素化和降解
6、SCD1抑制剂与多纳非尼在肝癌中的协同作用
作者的上述数据表明,抑制SCD1可显著逆转肝癌细胞对TKIs的耐药性。作者推测TKIs和SCD1抑制剂联合使用可能对p53野生型癌症有效。Aramchol是一种新型的SCD1部分抑制剂,在一项2b期临床试验中,与其他SCD1抑制剂相比,Aramchol已被证明是非酒精性脂肪性肝炎(NASH)的一种有希望的治疗方法,且未导致严重不良事件。为了评估aramchol在体内的安全性和抗肿瘤活性,作者使用了啮齿动物的异种移植模型。结果表明aramchol在小鼠模型中的安全性。
Aramchol在体外对细胞活力没有明显的抑制作用(图6a),与异种移植模型的结果一致。值得注意的是,在长期克隆实验中,aramchol增强了p53野生型癌细胞对多纳非尼的敏感性,多纳非尼是一种氘化的索拉非尼衍生物,比索拉非尼更有优势(图6b, c)。作者接下来研究了aramchol是否可以在HCC患者衍生的类器官(PDOs)中与多纳非尼协同作用。作者也确定了PDOs中p53的状态(图6d)。与癌细胞系的结果相似,URI缺失显著提高了多纳非尼在p53野生型PDOs中的敏感性。多纳非尼和aramchol联合使用确实比单独使用多纳非尼表现出更强的细胞毒性作用(图6e-g)。此外,与单一药物相比,联合治疗刺激了更高水平的脂质过氧化,这表明aramchol在临床前模型中促进了多纳非尼诱导的铁死亡(图6h)。相比之下,p53突变的PDOs对多纳非尼的耐药性更强。
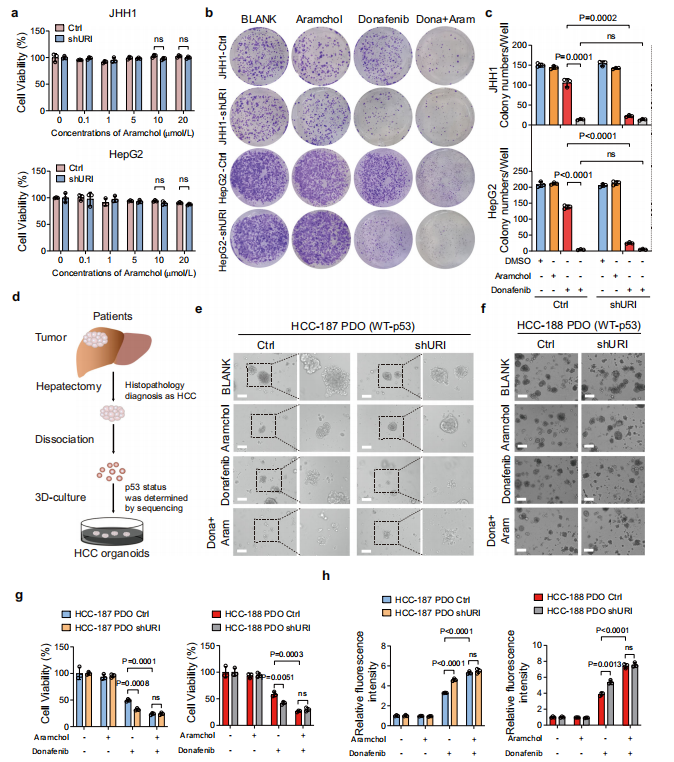
图6 SCD1抑制剂芳烃可增强多纳非尼在癌细胞和患者源性类器官中的疗效
作者进一步探讨了联合治疗在体内的治疗效果(图7a)。在HepG2-Ctrl和HepG2-shURI细胞异种移植的小鼠中,aramchol和多纳非尼联合使用可完全抑制肿瘤生长,而多纳非尼单药治疗HepG2-shURI细胞的异种移植具有有效的治疗作用(图7b-d)。肿瘤切片的免疫组织化学化学分析进一步显示,aramchol和多纳非尼治疗小鼠的肿瘤中Ki67降低。此外,研究发现,aramchol和多纳非尼联合使用可促进SCD1和p53的协同丧失(图7e, f)。综上所述,这些结果表明SCD1抑制剂aramchol与多纳非尼对肝癌具有协同致死性。
考虑到临床上TKIs主要用于晚期HCC患者。作者延迟给药,以使HepG2异种移植物的肿瘤生长到适当的大小(图7g)。再一次,联合治疗引起了明显的肿瘤控制(图7h, i)。此外,联合治疗显著提高了肿瘤中的脂质过氧化水平(图7j)。因此,多纳非尼和aramchol联合使用可能会获得显著的临床益处,特别是对于野生型p53的HCC患者。
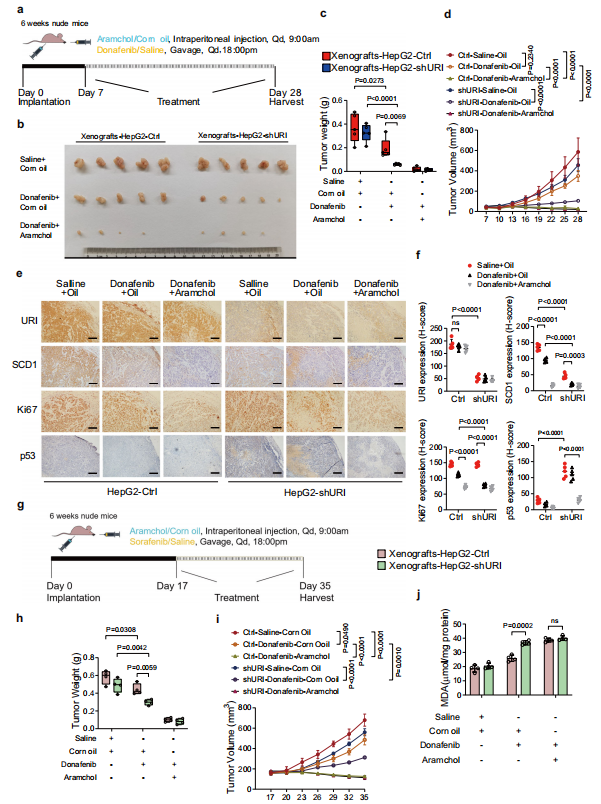
图7 SCD1抑制剂芳烃可增强多纳非尼的体内疗效
7、URI和SCD1的高表达与HCC预后恶化和索拉非尼耐药相关
在作者之前的研究中,作者发现URI在HCC中高表达与肿瘤恶性和患者生存率低相关。为了进一步证实URI-SCD1轴与临床癌症治疗之间的功能联系,作者首先利用组织显微阵列,包括134例晚期HCC样本(队列A)。作者在这些样本中进行多重免疫组织化学/免疫荧光(mIHC/IF)检测URI和SCD1表达。采用H-score法定量表达,见材料和方法(图8a)。在队列A样本中,URI表达与SCD1表达呈正相关(图8b, c)。此外,作者根据URI和SCD1的表达将患者分为四组。通过Kaplan-Meier曲线(p < 0.0001, log-rank检验)显示不同亚类间OS的显著差异(图8d)。URI和SCD1协同表达较高的患者风险最高,5年OS率为4.3% (HR, 2.65;95% CI, 1.76-3.98)而低URI和SCD1的患者为18.8%。这些结果表明URI和SCD1的联合表达是HCC预后不良的一个非常有效的预测因子。
由于作者的队列A中没有p53状态数据,为了进一步研究URI-SCD1在不同p53状态HCC患者中的作用,作者采用了Gao等人招募的一个新的HCC队列,作者将其命名为“Fudan_HCC_cohort”。通过分析该队列的WES和转录组数据,作者发现在p53-WT状态的HCC患者中,URIlow肿瘤中的SCD1表达低于URIhigh肿瘤,而其他与铁死亡相关的分子,如ACSL4、ALOX12、GPX4、SLC7A11和AIFM2没有明显改变(图8e)。此外,p53- WT型HCC患者中URI或SCD1的高表达与较差的临床预后相关(图8f, g)。综上所述,这些结果表明,在野生型p53患者中存在URI-SCD1与HCC患者临床预后之间的潜在相关性,而在p53突变型HCC患者中则不存在。
为了进一步探讨URI-SCD1在索拉非尼耐药中的作用是否与临床相关,作者在接受索拉非尼辅助治疗的HCC患者队列(队列B)中检测了URI和SCD1的表达。作者的数据显示URI表达与SCD1表达密切相关,与队列A的结果一致
然后,作者使用之前的队列(命名为C队列),该队列纳入了复发性HCC患者,然后患者接受含有索拉非尼的全身治疗。45例患者为p53- WT, 1例患者携带p53同义体突变,作者也将其分组为p53- WT。免疫组化法检测SCD1、URI、p53蛋白表达水平。该队列中p53-WT患者的总生存率高于p53突变患者(图8h)。作者发现在p53-WT组中SCD1与URI之间存在显著相关性,而在p53-突变组中则没有(图8i-k)。同时,在接受索拉非尼治疗的p53-WT HCC患者中,较高水平的SCD1或URI与预后恶化相关(图8i - o)。因此,作者的研究结果证明了URI-SCD1轴在p53-WT HCC患者索拉非尼耐药中的重要作用。
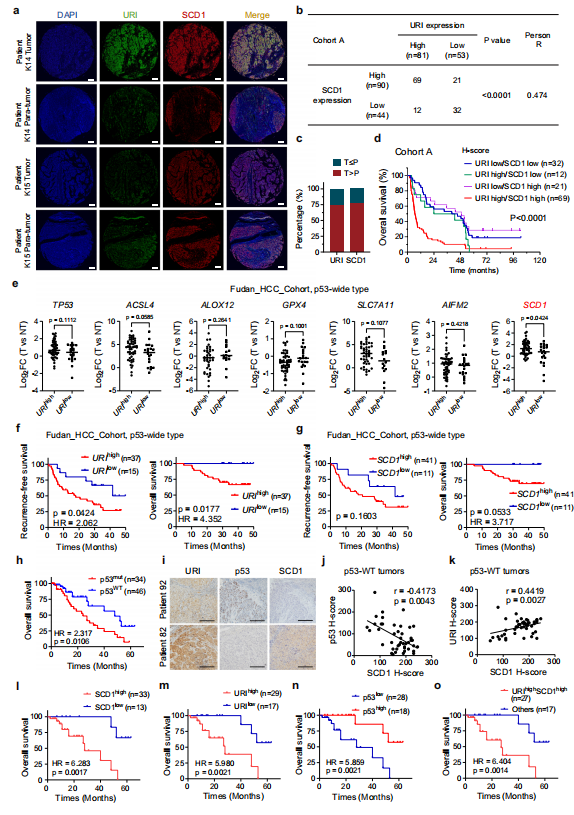
图8 URI联合SCD1与晚期HCC患者生存差和索拉非尼耐药相关
结论
综上所述,URI以TRIM28-MDM2依赖的方式维持低水平的p53,维持SCD1活性和MUFAs的积累,从而促进癌细胞对TKIs的耐药性。URI-p53-SCD1轴介导TKIs的耐药,这可能解释了为什么p53野生型HCC仍然对TKIs表现出内在耐药。此外,本研究确定的联合治疗可能代表了约41%具有野生型p53和高水平URI/SCD1的晚期HCC患者的一种有希望的策略(图9)。本研究为作者后续的临床试验提供了理论基础。
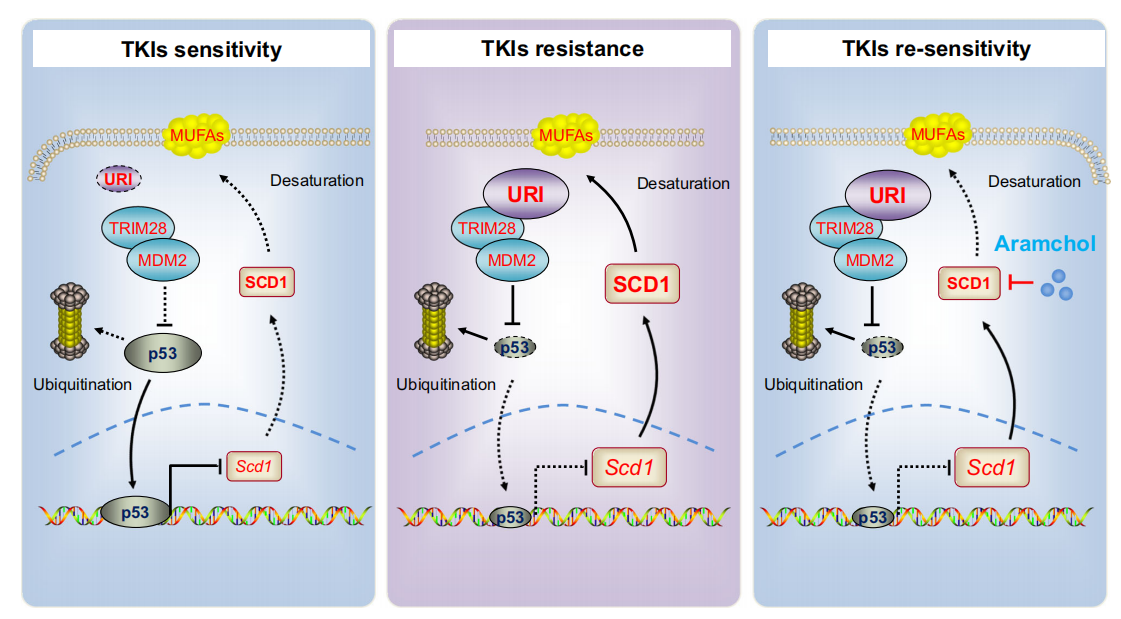
图9 URI-p53-SCD1轴介导TKIs耐药和芳烃再敏化肝癌细胞对TKIs的模型
实验方法
细胞系和细胞培养;人肝细胞癌衍生类器官的培养;实时荧光定量PCR分析;脂质过氧化实验;细胞增殖、菌落形成和活力测定;流式细胞术;WB、免疫共沉淀和抗体;荧光素酶报告实验;染色质免疫共沉淀;肿瘤异种移植实验;免疫组织化学和评分;多路复用免疫组织化学和免疫荧光;RNA测序;CUT&Tag库建设;LC-MS;靶向中、长链脂肪酸定量;蛋白LC-MS/MS分析;DNA电泳;重组Myc-URI和His-TRIM28的生产。
参考文献
Ding Z, Pan Y, Shang T, Jiang T, Lin Y, Yang C, Pang S, Cui X, Wang Y, Feng XF, Xu M, Pei M, Chen Y, Li X, Ding J, Tan Y, Wang H, Dong L, Wang L. URI alleviates tyrosine kinase inhibitors-induced ferroptosis by reprogramming lipid metabolism in p53 wild-type liver cancers. Nat Commun. 2023 Oct 7;14(1):6269. doi: 10.1038/s41467-023-41852-z. PMID: 37805657; PMCID: PMC10560259.