






基于小鼠结直肠癌模型和类器官的单细胞转录组分析揭示晚期结直肠癌治疗新靶点

结直肠癌(CRC)是癌症相关死亡的第三大原因,大多数(>80%)CRC是由肿瘤抑制因子APC的功能丧失(LOF)突变驱动的,导致典型WNT途径效应因子β-连环蛋白的组成性激活。本文通过在对APC失活的结肠类器官进行转录组分析,并将基因表达变化与几种结肠癌小鼠模型(包括炎症诱导的CRC模型)的转录组进行了交叉参考,发现了棕榈油酰-β蛋白羧酸酯酶(Notum)在APC损失时被急性诱导并且在这些模型中保持升高,采用体外测定、体内内窥镜引导的原位类器官植入测定和转录组学分析来表征Notum活性的作用。发现了靶向细胞外酶Notum的单一药剂在治疗临床前小鼠模型和人类类器官中的高度侵袭性转移性腺癌中的重要作用。本文于2023年8月17日发表于《gut》上,IF=24.5。
技术路线






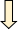

主要研究内容
1.进展期结直肠腺癌中NOTUM的致癌活性
在APC失活后的结肠类器官进行了转录组分析,并对几种结肠癌小鼠模型的转录组进行了交叉参考基因表达变化(图1A)。在这些模型中表达在APC损失时被急性诱导并且保持升高的大约150个基因中,棕榈油酰基-α蛋白羧酸酯酶Notum脱颖而出,因为它先前涉及腺瘤形成的起始。在小鼠肿瘤中证实了Notum的高表达,其中其表达限于上皮/肿瘤细胞(图1B)。还证实了Notum对体内结肠上皮中急性遗传消融Apc或β-连环蛋白激活的强烈激活。此外,相对于正常组织,在人癌症基因组Atlas 3中的结肠癌和直肠癌(分别为TCGA结肠腺癌(COAD)和直肠腺癌(READ))中NOTUM表达显著升高(图1C)。同时,也检查了原代人COAD 的单细胞转录组谱,并证实NOTUM表达主要限于癌细胞,并且几乎完全不存在于肿瘤微环境中(图1D)。最初评估了NOTUM在具有侵袭性和转移能力的COAD小鼠模型中的功能:在引入靶向Notum的gRNA时,类肿瘤培养物生长较慢,并且具有显著降低的克隆类肿瘤接种效率(单细胞接种新类肿瘤的能力,代表肿瘤启动能力)(图1E-H)。为了测试NOTUM在体内是否具有相似的致癌活性,用APKS类肿瘤和NOTUM LOF进行了皮下和原位肿瘤形成测定。这些测定揭示了NOTUM LOF导致原发性肿瘤生长的显著降低,并且在原位模型中,肝脏中的大转移的完全阻断(图1 I、J)。这些结果揭示了NOTUM在CRC中的致癌活性。
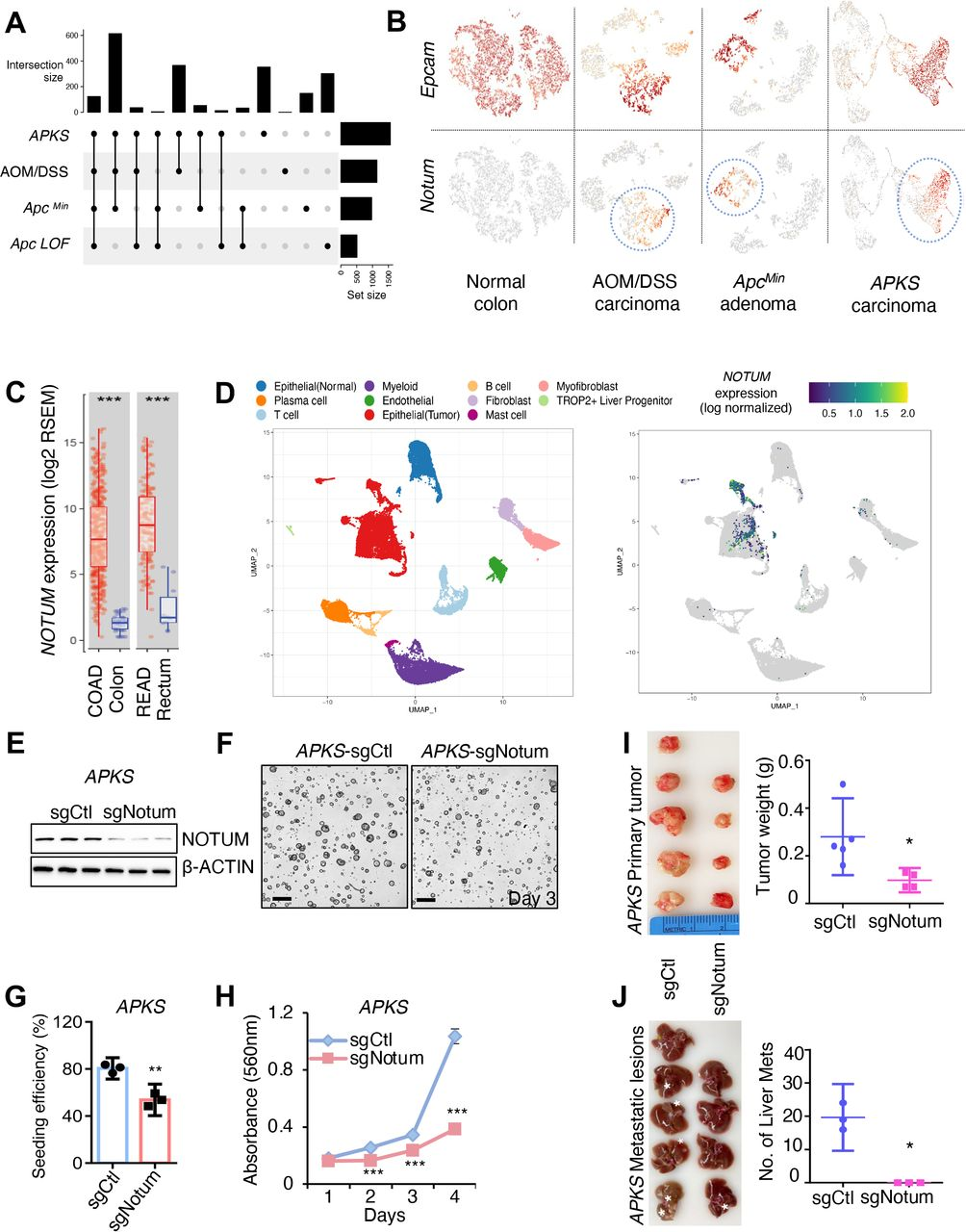
图1:进展期结直肠腺癌中NOTUM的致癌活性
2.NOTUM维持对APC失活的肿瘤抑制活性
通过CRISPR/Cas9在大量野生型结肠类器官培养中的NOTUM LOF导致克隆类器官接种效率、类器官生长和β-连环蛋白靶基因表达的显著增加(图2A-C),并且NOTUM过表达具有相反的作用(图2D、E),表明NOTUM负调节正常肠干细胞区室中的经典WNT途径。在具有由CRISPR/ Cas9(ApcΔ)引入的失活Apc突变的腺瘤类肿瘤中进行了类似的实验。值得注意的是,ApcΔ 类肿瘤中的NOTUM GOF导致克隆接种效率和增殖降低,并且NOTUM LOF具有相反的作用(图2F-K)。因此,想要探讨NOTUM丢失如何影响ApcΔ类肿瘤在体内的发生。内窥镜引导的ApcΔ肿瘤样细胞原位植入证实,NOTUM在不存在APC功能的情况下保留了有效的肿瘤抑制活性,ApcΔ肿瘤样细胞仅在结肠上皮中形成小腺瘤。相比之下,NOTUM的丢失显著增加原发性肿瘤大小并促进转移(图2L-N)。因此,NOTUM在APC失活驱动的腺瘤性病变中保留了有效的肿瘤抑制活性。
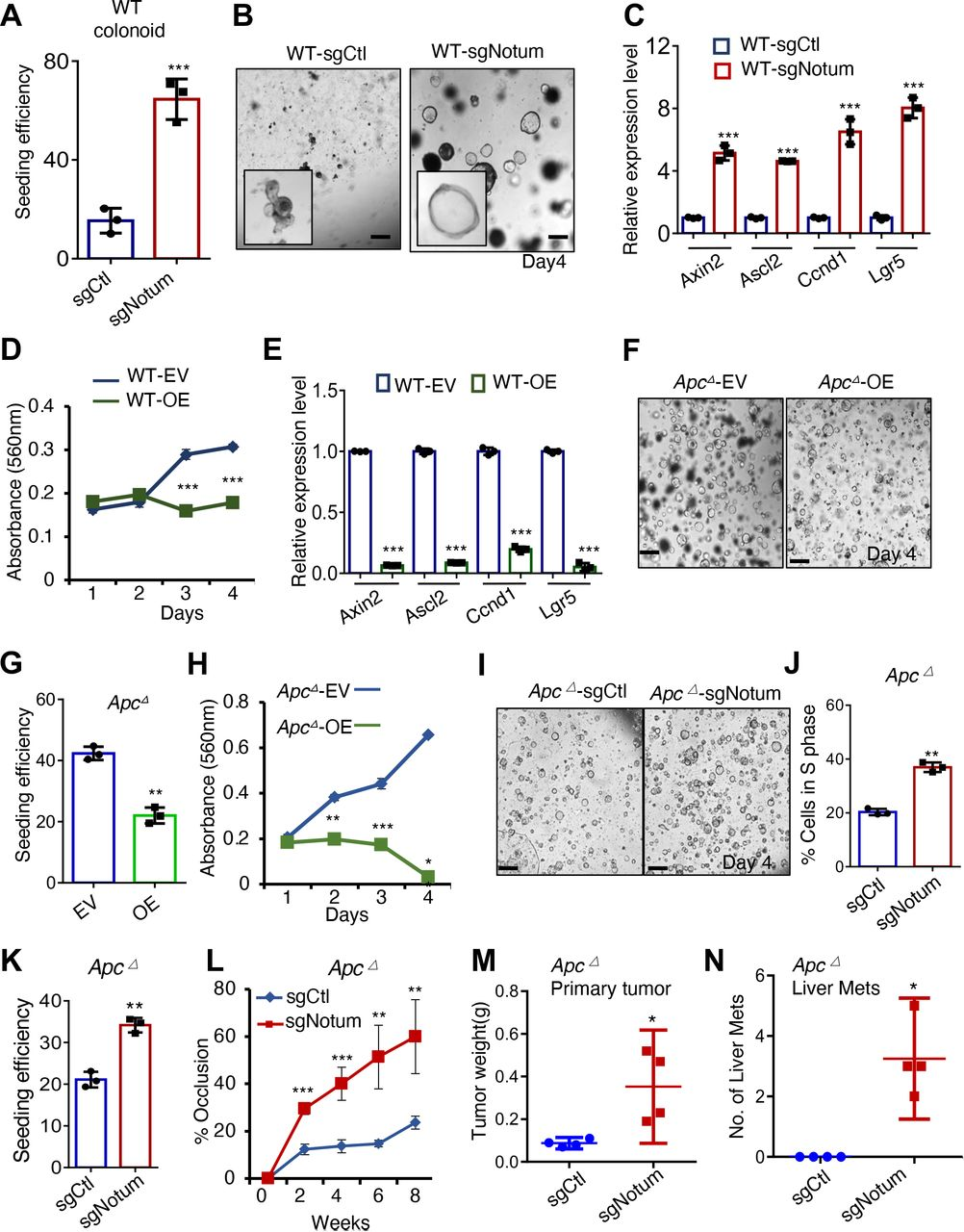
图2:NOTUM在正常和APC突变小鼠结肠类肿瘤中具有强效的肿瘤抑制活性
3.NOTUM的新的肿瘤抑制和致癌功能需要细胞外酶活性
由于在ApcΔ类肿瘤和更具侵袭性的AP/APK/APKS类肿瘤中的发现基于哺乳动物中NOTUM功能模型是出乎意料的,因此接下来测试了NOTUM在Apc Δ类肿瘤中的肿瘤抑制活性和AP/APK/APKS类肿瘤中的致癌活性是否需要酶活性。我们在小鼠Apc Δ类肿瘤中过表达野生型或催化死亡(S239 A)NOTUM,并且发现NOTUM S239A不能抑制增殖和克隆接种效率(图3A-C )。相反的,在亚纯型APKS亚克隆中过表达野生型和S239A NOTUM,并且发现,虽然野生型NOTUM增加类肿瘤接种效率和增殖,但催化死亡的NOTUM没有增加(图3D-F)。因此,催化活性对于NOTUM在ApcΔ类肿瘤中的肿瘤抑制活性和APKS类肿瘤中的致癌活性都是必需的。在Notum亚型APKS类肿瘤中用mCherry标记一半的培养物,另一半用表达GFP和NOTUM或仅表达GFP的载体标记。与ApcΔ类肿瘤一样,我们发现GFP+,NOTUM过表达细胞能够驱动mCherry+细胞非-细胞自主增殖,以及GFP+APKS细胞的细胞-细胞自主增殖(图3G,H)。因此得出结论,NOTUM的新致癌活性需要催化活性,并发生在细胞外。
图3:NOTUM的肿瘤抑制和致癌功能需要催化活性
4.Apc和Trp53失活协同赋予NOTUM致癌活性
已经确定NOTUM酶活性在野生型和Apc∆类肿瘤中具有肿瘤抑制性,并且在AP/APK/APKS类肿瘤中具有致癌性,因此推断基因开关必须是NOTUM功能逆转的基础,并且P53失活可能是原因。Notum表达在Apc失活时强烈上调,并且在致癌Trp53、Kras和Smad 4突变积累时保持升高(图4A)。相比之下,单独失活Trp53对Notum表达几乎没有影响。通过测定Notum LOF和GOF在Apc∆、Trp53∆和AP类肿瘤中的作用,并且显著地发现,在Apc∆和Trp53∆类肿瘤中,NOTUM活性保持肿瘤抑制性,但是这些突变一起协同作用以赋予NOTUM致癌活性(图4B-F)。通过收集来自COAD和来自TCGA的READ的患者数据,并基于具有APC突变但野生型TP53的患者或携带APC和TP53突变的患者中的NOTUM表达对患者无疾病生存期进行分层。与实验发现一致,高NOTUM表达预测仅在TP53突变型肿瘤中无疾病存活的显著降低,而在TP53野生型肿瘤中无疾病存活的显著降低(图4G)。为了描绘NOTUM的肿瘤抑制性到致癌转换的分子机制,进行了具有或不具有NOTUM LOF的ApcΔ和AP培养物的转录组谱分析(图4 H)。在ApcΔ和AP突变体培养物之间受NOTUM LOF影响的通路包括mTORC1和E2F信号传导(在ApcΔ中由NOTUM LOF诱导,而不是AP)、干扰素α/γ信号传导(在AP中由NOTUM LOF诱导,在AP中抑制)和TGF β(在AP中由NOTUM LOF诱导,而不是在Apc Δ中由NOTUM LOF诱导)(图4 I)。总之,这些结果表明了Apc和Trp53失活协同赋予NOTUM致癌活性。

图4:APC和P53丢失协同作用将NOTUM从肿瘤抑制因子转化为癌蛋白
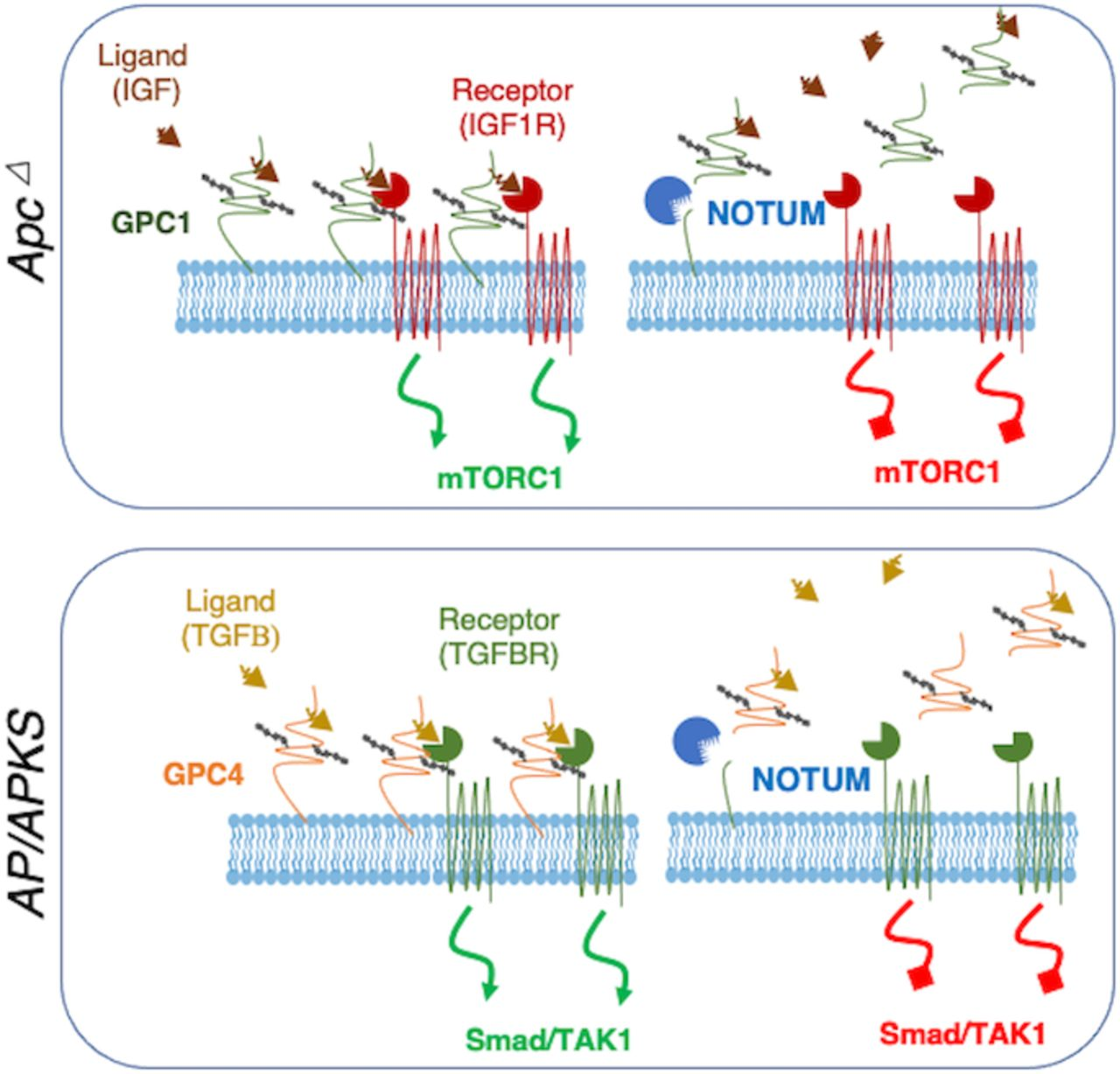
5.NOTUM的肿瘤抑制功能与致癌功能可以通过GPC活性差异来解释
为了探究磷脂酰肌醇蛋白聚糖Gpc 1/4基因失活是否会影响表型NOTUM活性。GPC4仅在AP类肿瘤中显示肿瘤抑制活性,在Apc Δ类肿瘤中没有作用(图5A-B)。表达低但可检测的Gpc 6在任一基因型中几乎没有影响或没有影响(图5A-B)。为了将NOTUM功能、GPC蛋白和信号传导通路活性联系起来,敲低了小鼠ApcΔ和AP类肿瘤中的Notum,并检查通路活性和细胞相关的GPC蛋白水平。Apc Δ 类肿瘤中的Notum敲低导致mTORC1信号传导增加,与GPC1蛋白的积累偶联,对TGF β途径活性没有明显变化(图5C)。相比之下,AP类肿瘤中Notum敲低导致TGF β途径活性增加和GPC4蛋白积累,而对mTORC1活性没有明显影响(图5D)。这些实验测定了全细胞裂解物中的GPC蛋白水平,并支持一种模型,其中Notum LOF阻止GPC从细胞表面切割导致其积累。为了进一步探索这种可能性,检测了Notum抑制后Apc Δ和AP类肿瘤的培养基上清液中游离N-β末端GPC1和GPC4的水平。相对于上清液中的GPC1,ApcΔ类肿瘤中的NOTUM LOF增加了细胞相关GPC1(图5E)。相反,AP类肿瘤中的Notum LOF导致细胞相关GPC4增加,同时上清液中GPC4减少(图5E)。有趣的是,NOTUM蛋白本身与细胞裂解物而不是上清液相关联,这一发现与NOTUM建立的胞外酶促作用相结合,表明NOTUM与胞外细胞表面物理相关联(图5E)。为了进一步测试GPC操作的效果是否与NOTUM分别从Apc Δ和AP类肿瘤表面切割GPC1和4一致,分析了GPC失活对受NOTUM活性差异影响的途径的影响。事实上,小鼠Apc∆类肿瘤中的GPC 1失活抑制mTORC1活性,小鼠AP类肿瘤中的GPC 4失活抑制TGF β活性,与NOTUM操作的效果一致(图5F-G)。最后,为了确认GPC1或GPC4和受影响的信号转导途径上游的受体之间的直接相互作用。GPC1在Apc Δ类肿瘤中的免疫沉淀共免疫沉淀IGF1Rβ,一种与结肠癌和化疗耐药性有关的mTORC 1活化上游的IGF受体(图5 H)。在AP类肿瘤中,GPC4的免疫共沉淀TGF β通路上游的TGFβR1(图5I)。添加小分子NOTUM抑制剂ABC 99似乎增强了这些相互作用(图5 H-I)。因此,研究了NOTUM对通过TAK1-p38 a信号转导介导的非典型TGFβ途径活性的影响。与在AP类肿瘤中的发现一致,NOTUM抑制导致APKS细胞裂解物中GPC4增加(图5 J)。进一步与NOTUM作为TGFβ信号传导负调节剂的作用一致,APKS类肿瘤中的Notum抑制导致TGFβ活化激酶(TAK 1)的磷酸化和下游p38a磷酸化增加(图5 J)。观察到AP和APKS类肿瘤中的p38a抑制增加增殖并抑制细胞死亡(图5 K)。这些发现支持了NOTUM可能在腺癌中发挥致癌作用的观点,至少部分是通过抑制非α经典TGFβ途径的肿瘤抑制活性。

图5:磷脂酰肌醇蛋白聚糖介导Apc突变体与Apc/Trp 53突变体小鼠类肿瘤中NOTUM活性的差异效应
6.药理学NOTUM抑制在转移性结肠癌的临床前动物模型中有效
最初证实ABC99处理表型模仿了Apc∆和APKS类肿瘤中NOTUM的遗传抑制(图6A和B)。接下来,将APKS类瘤通过内窥镜引导的注射将原位植入到同系小鼠的结肠粘膜中,并允许肿瘤移植并生长1个月,之后将小鼠随机分配至媒介物对照或ABC 99组。然后用ABC 99或媒介物对照再处理小鼠4周。ABC 99治疗在很大程度上阻止了原发性肿瘤生长,BLI和肿瘤重量相对于对照动物显著降低(图6C-E)。值得注意的是,NOTUM抑制几乎完全抑制了该高度侵袭性CRC模型中的肝脏和淋巴结转移(图6 F和G),并且ABC 99-γ处理的原发性和罕见转移性病变在实验结束时表现出显著的细胞增殖抑制(图6 H)。构建了另一组具有原位APKS肿瘤的小鼠,如前所述允许它们移植1个月,然后开始ABC99或媒介物对照治疗并监测存活长达4个月。值得注意的是,虽然媒介物对照组-β处理的小鼠在该时间过程中变得垂死,但ABC99-β处理的组在该实验的持续时间内均未死于疾病(图6 I)。
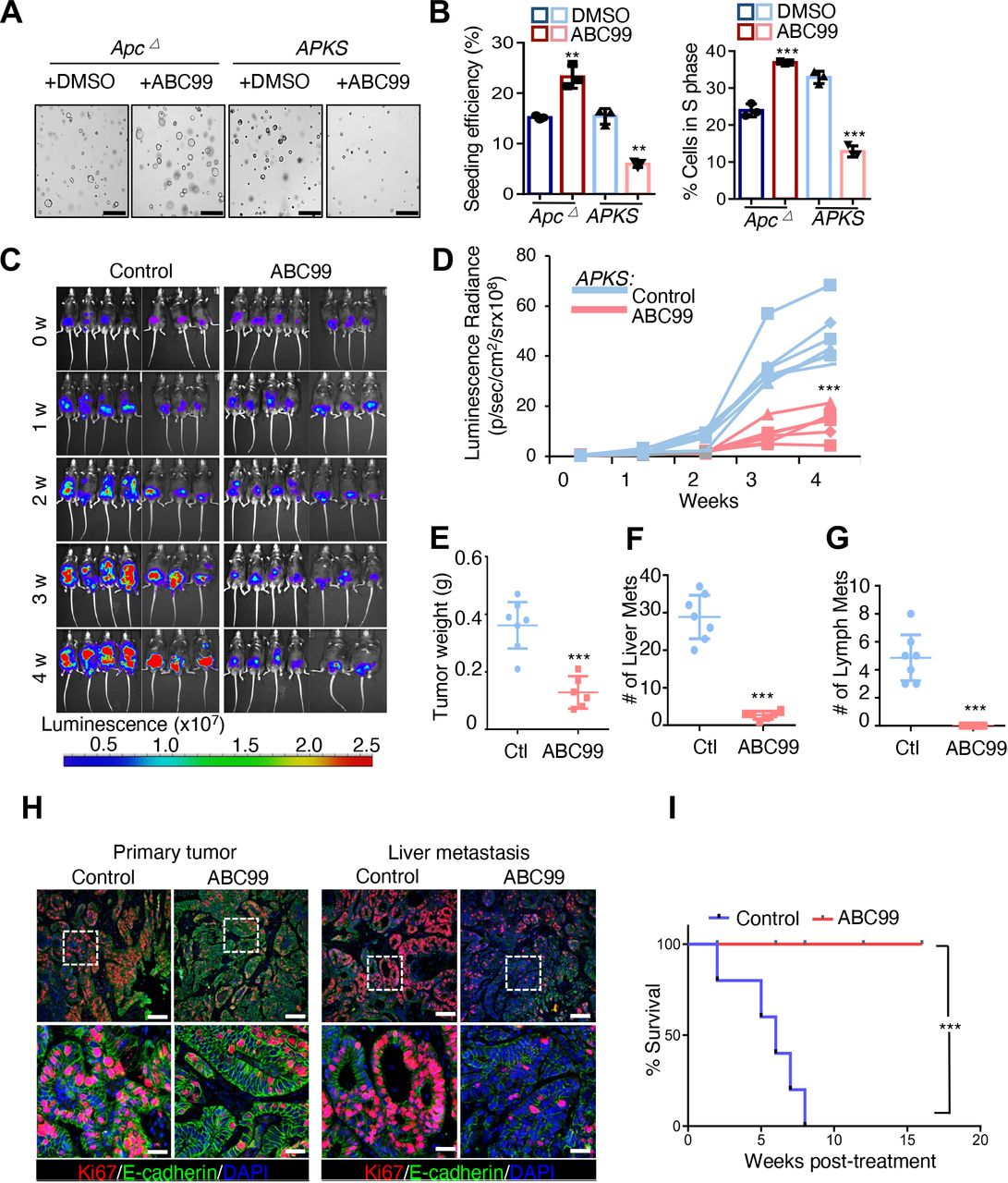
图6:NOTUM的小分子抑制剂抑制结直肠癌小鼠模型中的肿瘤生长和转移
结论
本研究发现了NOTUM在APC缺失腺瘤中保留肿瘤抑制活性,另外,在进展为腺癌伴P53丢失时,NOTUM成为专性癌基因。这些表型是不依赖于Wnt-β的,分别由NOTUM在早期阶段与晚期阶段疾病中对Gpc1和4的不同活性引起。最终,临床前小鼠模型和人类器官培养物证明,NOTUM的药理学抑制在阻止原发性腺癌生长和抑制远端器官的转移性定殖方面是高度有效的。因此,靶向细胞外酶NOTUM的单一药物在治疗临床前小鼠模型和人类类器官中的高度侵袭性转移性腺癌中是有效的,这使得NOTUM及其磷脂酰肌醇聚糖靶向晚期CRC中的治疗弱点。
实验方法
构建正常和肿瘤类器官,AOM/DSS结直肠癌模型,ApcMin/+小鼠模型,单细胞转录组测序,原位移植瘤模型,原发肿瘤模型,Westernblot,MTT增殖实验,慢病毒感染,免疫荧光实验,流式细胞术,免疫共沉淀实验,生物发光成像实验,生存分析。
参考文献
Tian, Y., Wang, X., Cramer, Z., Rhoades, J., Estep, K. N., Ma, X., Adams-Tzivelekidis, S., Katona, B. W., Johnson, F. B., Yu, Z., Blanco, M. A., Lengner, C. J., & Li, N. (2023). APC and P53 mutations synergise to create a therapeutic vulnerability to NOTUM inhibition in advanced colorectal cancer. Gut, gutjnl-2022-329140. Advance online publication. https://doi.org/10.1136/gutjnl-2022-329140