






组蛋白乳酸化促进心肌梗死后修复性基因激活
心肌梗死(MI)后炎症的消退和心脏修复的启动需要及时激活修复信号。组蛋白乳酸化通过转录调控赋予巨噬细胞稳态基因表达特征。但组蛋白乳酸化在心肌梗死后修复反应中的作用尚不清楚。我们旨在研究组蛋白乳酸化是否在心肌梗死早期和远程后诱导单核细胞的修复性基因表达。我们证明组蛋白乳酸化通过促进修复基因转录调节单核-巨噬细胞的抗炎和促血管生成双重活性,并证实组蛋白乳酸化有利于修复环境并改善心肌梗死后的心功能。此外,我们探索了单核细胞组蛋白乳酸化在再灌注心肌梗死中的潜在积极作用。机制上,我们提供了新的证据,证明单核细胞在心肌梗死早期经历代谢重编程,并证明糖酵解和MCT1(单羧酸转运蛋白1)介导的乳酸转运失调促进组蛋白乳酸化。最后,我们揭示了IL(白细胞介素)-1β依赖的GCN5(一般控制非抑制性5)募集对组蛋白H3K18乳酸化的催化作用,并阐明了其作为上游调控元件在单核细胞组蛋白乳酸化调控和心肌梗死后下游修复基因表达中的潜在作用。组蛋白乳酸化促进单核细胞修复转录反应的早期远程激活,这对于心肌梗死后免疫稳态的建立和心脏修复过程的及时激活至关重要。本文于2022年11月发表于Circulation Research (IF=23.213)。
技术路线

结果
(1) 修复基因在心脏招募前在骨髓和循环单核细胞中被早期和远程激活
单核巨噬细胞从炎症状态及时过渡到修复状态决定了免疫稳态和心肌梗死的结局。我们假设修复性基因激活可能在心脏招募前在骨髓和循环单核细胞内启动。为验证这一假设,使用不同时间点心肌梗死小鼠BM和循环白细胞的单细胞RNA-seq数据进行了生物信息学分析。在解复用和定位到参考基因组后,我们进行了粗略的聚类,以大致定义5种主要的细胞类型,并关注单核细胞上(图1A和1B)。在27284个单细胞转录组中,5346个单核细胞根据样本来源和心肌梗死后的不同时间点分为7组:心肌梗死后0、1、2天的BM单核细胞和心肌梗死后0、1、2、4天的外周血单核细胞(图1C)。单细胞转录组数据的基因本体学(GO)术语分析揭示了心肌梗死第一天单核细胞群体中显示的主要过程,表明单核细胞除了上调和放大心肌梗死后的炎症反应外,还迅速启动修复过程,包括对血管生成和伤口愈合的反应(图1D)。小提琴图显示了一系列经典修复基因,包括Anxa6、F10、Lrg1和Tgfbi在MI后早期的表达(图1E)。心肌梗死后单核细胞发育过程的伪时间轨迹分析显示,BM和循环单核细胞中的修复基因被早期激活,并随时间的推移逐渐积累(图1F和1G)。
为探讨心肌梗死后不同单核细胞亚群的修复转录反应,我们将单核细胞分为2个亚群;Ly6Chi和Ly6Clo,并检测了单细胞测序数据中不同亚群中代表性修复基因的表达(图S1E和S2G)。在第1天对假手术小鼠和心肌梗死后小鼠的循环Ly6Chi和Ly6Clo单核细胞进行分类后,采用逆转录-qPCR (RT-qPCR)检测修复基因的表达。修复基因在Ly6Chi单核细胞亚群和Ly6Clo单核细胞亚群中都在早期被激活(图S1F)。
为验证上述发现,对假手术或心肌梗死第1天小鼠的循环单核细胞进行了RNAseq分析,鉴定出心肌梗死后650个上调基因和203个下调基因,表明循环单核细胞在到达损伤心脏之前发生了早期和明显的转录组变化(图1H)。氧化石墨烯项富集分析证实,修复基因的表达,包括参与血管发育、伤口愈合和细胞外基质组织的基因,被广泛和远程激活(图1I)。为筛选以血管发生为代表的关键修复过程中的枢纽基因,使用CytoScape软件对血管发育中富集的基因进行了蛋白-蛋白相互作用网络分析。在基于MCC方法的网络中优先级最高的10个基因中,已知的修复基因Vegf-a和IL-10被确定为MI后早期修复网络的潜在枢纽基因(图1J)。此外,我们对心肌梗死和假手术小鼠术后第1天的BM单核细胞进行了RNA测序,心肌梗死后伤口愈合信号和相关修复基因的快速上调,证实了心肌梗死后单核细胞修复转录反应的早期远程激活(图S2H和S2I)。此外,BM和外周血中的Ly6Chi和Ly6Clo亚群都表现出早期修复途径的启动,包括血管生成、细胞外基质组装和IL(白细胞介素)-10的产生(图S2J至S2Q)。
总之,这些结果强调了单核细胞在转录水平上及时启动修复活性,并强调了通过早期对炎症和修复基因的细致调节,心肌梗死后免疫环境的动态调节。
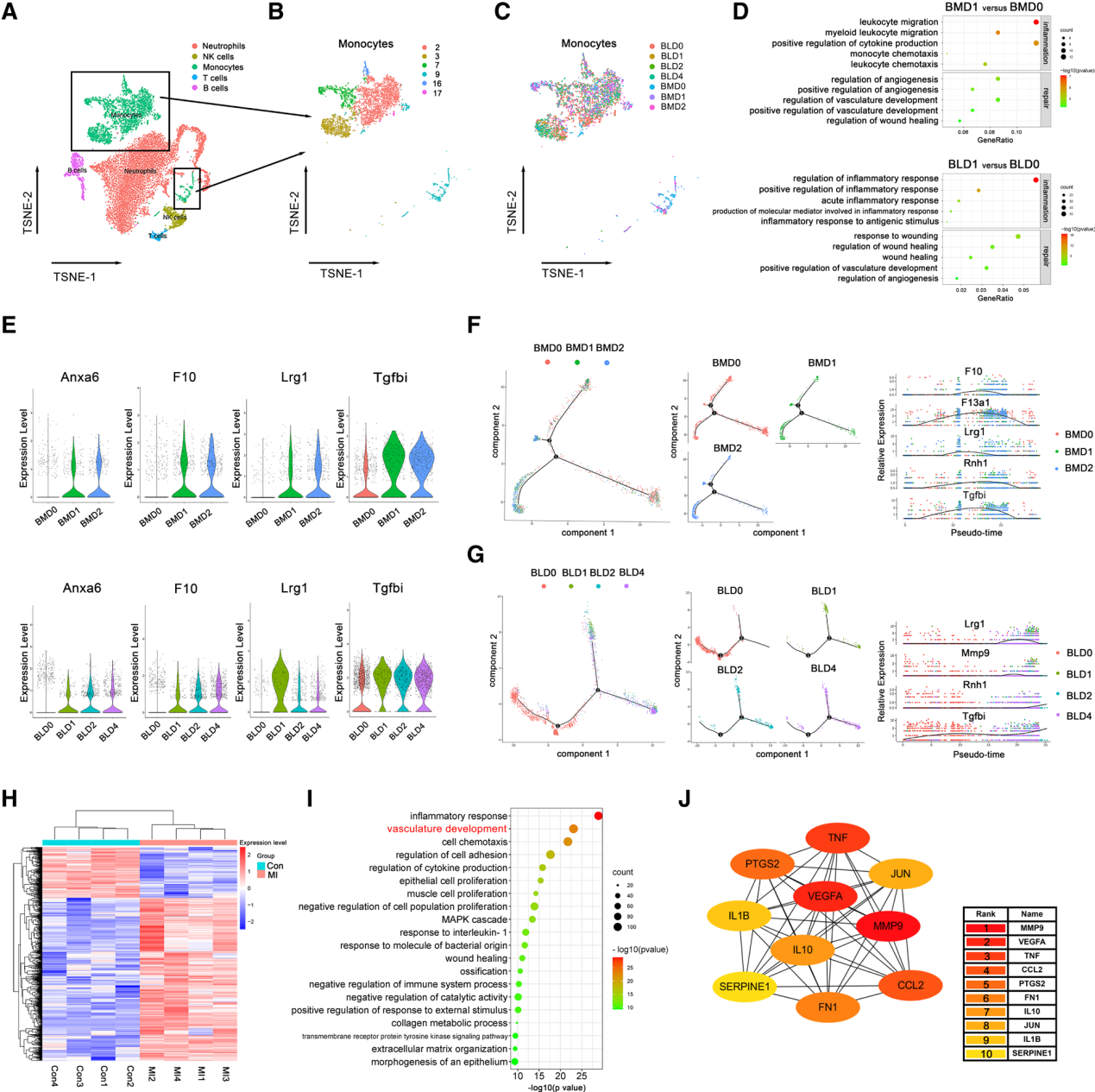
图1:心肌梗死后骨髓(BM)和循环单核细胞修复基因的早期激活
(2) 心肌梗死后骨髓和循环单核细胞组蛋白乳酸化迅速升高
为探讨乳酸化在心肌梗死后单核细胞修复基因的早期和远程激活中的作用,我们评估了心肌梗死早期心肌梗死中心肌和循环单核细胞以及心肌浸润巨噬细胞中乳酸化的变化。我们通过磁头分选从心肌梗死小鼠中分离心肌和循环单核细胞(图S3A和S3B)。WB分析显示,与对照组小鼠相比,MI小鼠骨髓和循环单核细胞的整体乳酸水平显著升高,并且在17 kDa附近出现了一个优势条带,这可能代表了组蛋白H3(图2A和2B)。在不同时间点采集心肌梗死后小鼠的单核细胞免疫印迹表明,乳酸化水平早在心肌梗死后4至24小时就显著上调,并保持高水平直至72小时。相反,乙酰化水平在心肌梗死早期增加,并在心肌梗死后72小时内迅速下降至基线水平(图2C至图2F)。我们检测了先前鉴定的组蛋白H3K18乳酸化水平(H3K18la),其趋势与全球乳酸化水平相似(图2G至2J)。H3K18la水平维持在高水平长达7天,并在心肌梗死后14天恢复到基线水平(图S3D)。同样,在Ly6Chi亚群中发现H3K18la水平上升,而Ly6Clo单核细胞也显示出类似上升趋势,但时间稍晚(图S3E和S3F)。与此一致,心肌梗死后3天的免疫荧光染色显示心肌梗死区整体乳酸化和H3K18la水平显著升高,合并共聚焦图像显示,乳酸化定位于梗死区巨噬细胞的细胞核(图2K和2L)。综上所述,心肌梗死后心肌梗死局部和远端外周的单核巨噬细胞显示组蛋白乳酸化增加。
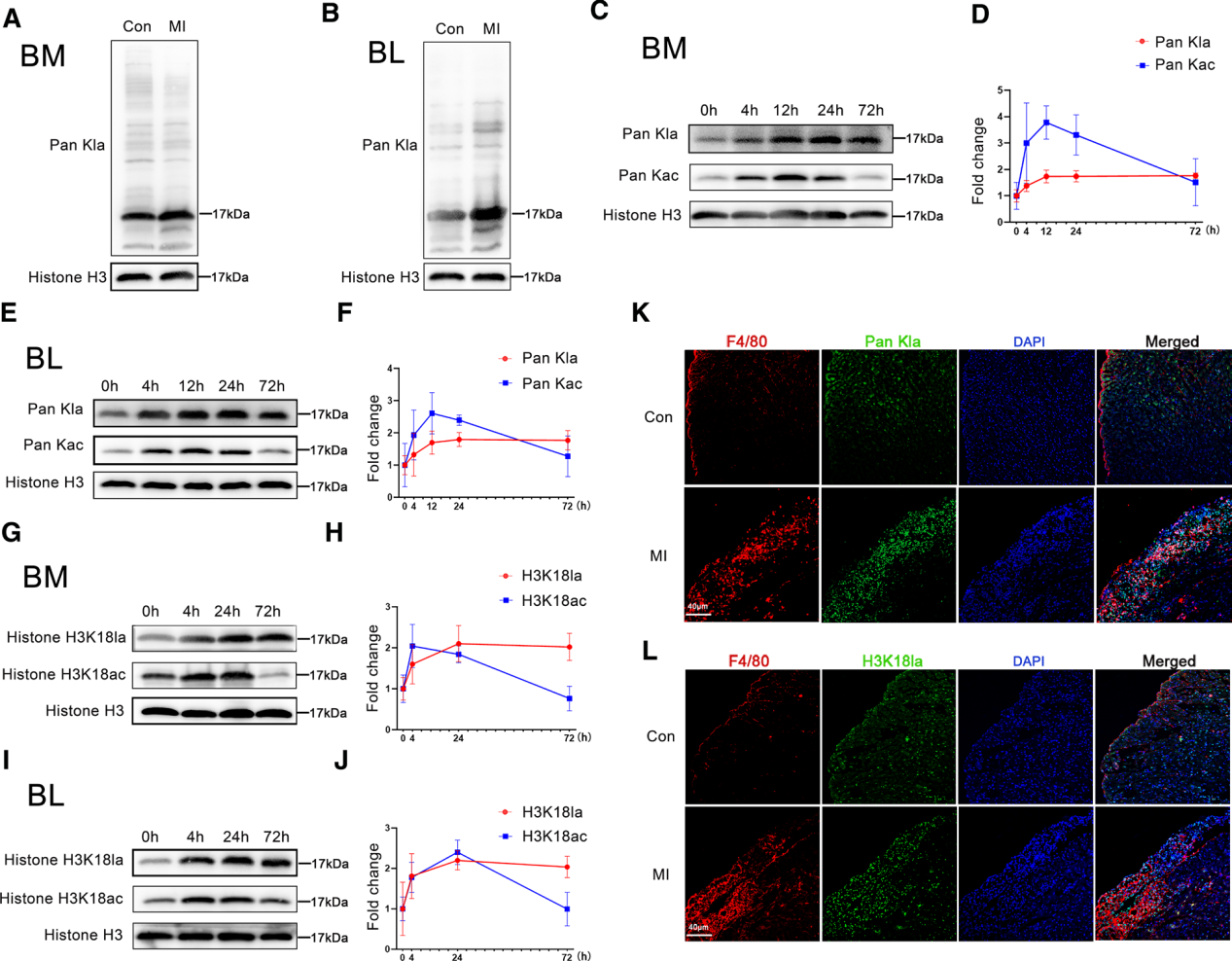
图2:心肌梗死(MI)后骨髓和循环单核细胞组蛋白乳酸化水平升高
(3) 组蛋白乳酸化促进修复基因Lrg1、Vegf-a和IL-10的激活
为筛选心肌梗死中组蛋白乳酸化的候选靶基因,从假手术小鼠和心肌梗死小鼠中筛选心肌梗死后24小时的循环单核细胞,并使用抗H3K18la或抗H3K18ac抗体对其进行CUT&Tag检测。H3K18la富集于基因的启动子区域和上游区域(图3A)。大多数H3K18la或H3K18ac增加的基因都是特异性的,H3K18la修饰相关基因与H3K18ac在心肌梗死后显著不同。这些基因中只有14%(4168个中的566个)同时显示H3K18la和H3K18ac显著增加,而63%的H3K18la标记基因没有显示H3K18ac显著增加(图3B和3C)。H3K18la修饰的基因主要参与免疫、内分泌和循环系统;心血管疾病;以及脂质和碳水化合物代谢(图S4B)。基因集富集分析表明,H3K18la结合的基因在很大程度上与血管生成和伤口愈合有关(图3D)。
为鉴定单核细胞MI后H3K18la靶基因,我们结合CUT&Tag和RNA-seq数据,根据基因表达和H3K18la修饰将基因分为4类:H3K18la差异或上调基因,H3K18la差异或下调基因(图3E)。图S4C中的热图显示了H3K18la修饰基因与上调基因的相关性。对两类H3K18la修饰基因的从头基序搜索,H3K18la上调和下调的基因具有不同的富集DNA序列(图3F)。GO分析显示,心肌梗死期间H3K18la修饰增加的上调基因积极参与血管生成的正调控(图3G)。RNAseq显示,许多参与血管生成过程的代表性基因(如Vegf-a、Lrg1和IL-10,也是H3K18la修饰升高的基因)在心肌梗死后显著上调(图3H)。我们筛选了H3K18la修饰上调的前30个基因,并根据转录上调进行排名;Lrg1表达上调最显著(图3I)。因此Vegf-a、IL-10和Lrg1作为乳酸化的潜在靶基因(图3J)。基因组快照显示了Vegf-a、IL-10和Lrg1中的H3K18la修饰位点(图S4D)。为在MI后1日的循环单核细胞中验证CUT&Tag和RNA-seq数据,进行了RT-qPCR和染色质免疫沉淀-qPCR (ChIP-qPCR)分析,以确认Vegf-a、IL-10和Lrg1的启动子区表达增加和H3K18la富集(图3K和3L)。只有Vegf-a在心肌梗死后表现出不同的H3K18ac修饰水平。ChIP-qPCR证实,心肌梗死后循环单核细胞中Vegf-a的H3K18ac修饰位点与H3K18la不同(图S4F)。qRT-PCR和ChIP-qPCR也验证了心肌梗死后不同单核细胞亚群中H3K18la靶基因的上调和乳酸化水平(图S4G至S4J)。
综上所述,在心肌梗死后单核细胞中确定了H3K18la的靶基因,H3K18la可启动心肌梗死后早期远程修复基因激活,有望促进心肌梗死后心脏及时修复。

图3:H3K18la启动心肌梗死(MI)后修复基因Lrg1、Vegf-a和IL-10的表达
(4) H3K18la调控巨噬细胞靶基因的转录和修复活性
为研究H3K18la对修复基因表达的直接影响,我们通过外源性乳酸或乳酸脱氢酶抑制剂FX-11处理骨髓源性巨噬细胞(BMDM)的组蛋白乳酸化水平。钙黄素和碘化丙啶染色和免疫印迹分析显示,在浓度高达40 µM时,FX-11对BMDMs无明显的有害作用,对H3K18la有明显的抑制作用(图S5A和S5B)。脂多糖(LPS)刺激BMDMs导致H3K18la水平升高(图4A)。添加外源性乳酸后,细胞内乳酸水平和组蛋白H3K18la水平升高(图4B和4C),与以往研究一致。ChIP-qPCR显示靶基因启动子区H3K18la富集增加,RT-qPCR结果证实乳酸处理后靶基因表达上调(图4D和4E)。FX-11显著抑制BMDM细胞内乳酸和H3K18la水平(图4F和4G)。ChIP-qPCR和RT-qPCR显示,FX-11处理后H3K18la强度和靶基因表达降低(图4H和4I)。
我们分析了H3K18la对体外BMDM功能的影响。采集BMDM并用流式细胞术进行分析(图S5C)。促炎M1巨噬细胞的百分比在乳酸组显著降低,而在FX-11组增加(图4J和4K)。为排除乳酸诱导的H3K18la通过改变环境酸度影响巨噬细胞表型的可能性,对BMDM进行了乳酸钠处理。乳酸钠增加组蛋白乳酸化水平的程度与乳酸相似,但不影响巨噬细胞的炎症表型(图S5D和S5E)。
通过EdU标记、transwell、划伤/伤口迁移和管形成试验,评估了操纵H3K18la水平的单核细胞的促血管生成能力。小鼠主动脉内皮细胞在乳酸或FX-11刺激的BMDM条件培养基中培养24小时。经乳酸处理的BMDMs条件培养基与小鼠主动脉内皮细胞增殖(图4L)和向井区或创口区域迁移(图4M和4N)以及形成更有组织的分支和更长的管长(图4O),提示H3K18la升高的单核细胞具有增强的内皮细胞成管能力。
综上所述,H3K18la促进了Lrg1、Vegf-a和IL-10的转录,提高了BMDM的抗炎和促血管生成双重修复活性。
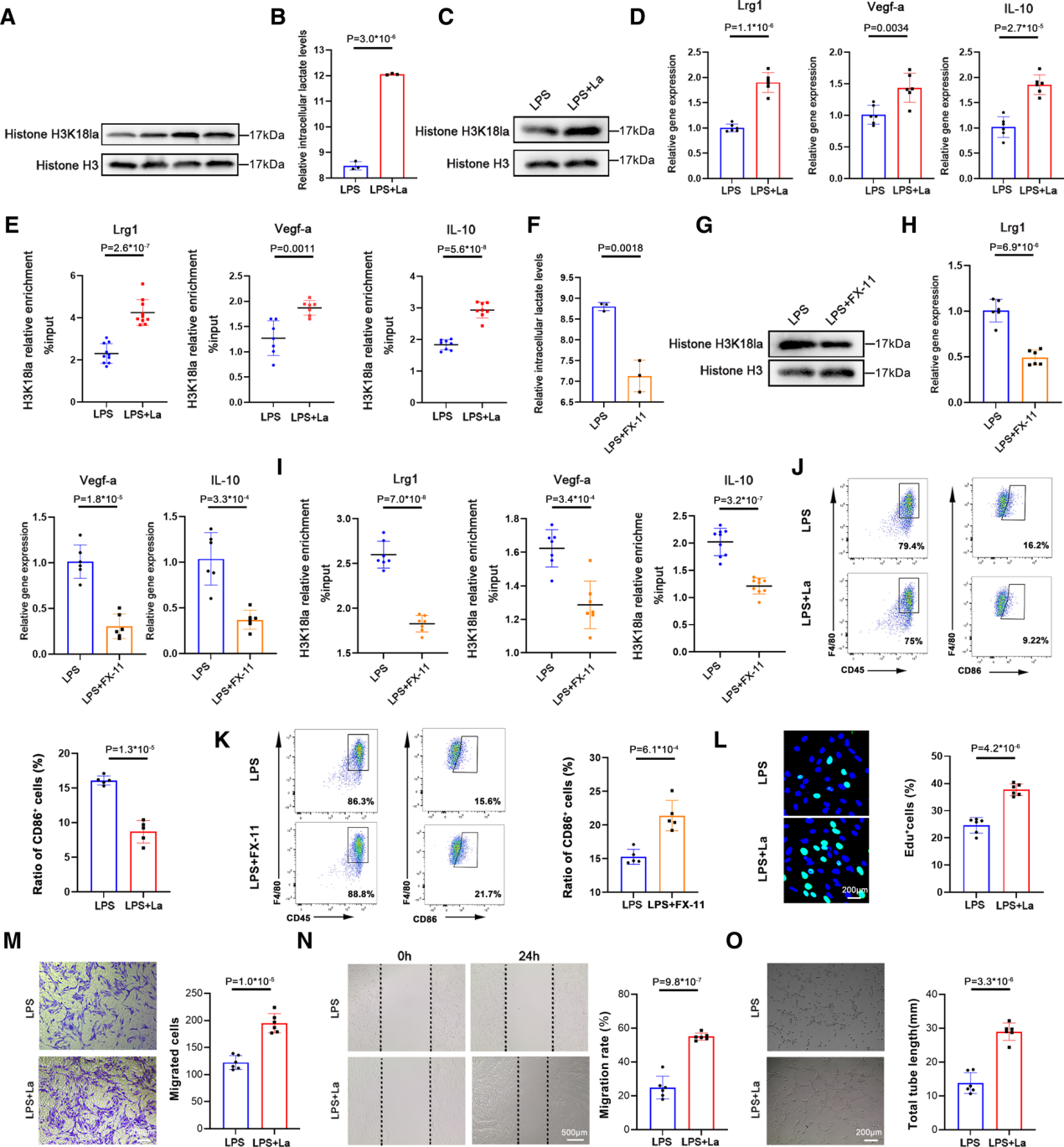
图4:组蛋白乳酸化激活靶基因转录和巨噬细胞修复活性
(5) 升高的组蛋白乳酸化有利于心肌梗死后的修复环境和改善心功能
为评估组蛋白乳酸化对缺血性心脏免疫环境的影响,我们建立了永恒性冠状动脉左前降支结扎的小鼠心肌梗死模型,并用乳酸钠或FX-11处理小鼠,以控制单核细胞组蛋白乳酸化。预先确定乳酸钠剂量,WB结果如图S6A和S6B所示。图5A为乳酸钠治疗心肌梗死小鼠的实验。乳酸钠处理小鼠循环单核细胞和浸润性巨噬细胞中的H3K18la水平显著升高(图5B和5C),Lrg1、Vegf-a和IL-10 mRNA水平升高(图5D)。
乳酸钠处理后H3K18la的增加抑制了炎症因子IL-6和TNF(肿瘤坏死因子)-α的表达(图5E)。流式细胞术显示,升高的H3K18la抑制了Ly6Chi炎性巨噬细胞和中性粒细胞的浸润(图5F和5G)。HE染色证实H3K18la升高导致炎症细胞浸润减少(图5H)。CD31和α-SMA (α-平滑肌肌动蛋白)的免疫荧光染色表明,乳酸钠诱导的H3K18la升高促进心肌梗死后血管生成(图5I和5J)。
马松三色染色显示,乳酸钠诱导的H3K18la升高可抑制心肌过度纤维化和病理性心脏重构(图5K)。乳酸钠也调节成纤维细胞的活化和胶原合成(图S7A至S7D)。超声心动图和磁共振显示,在基线时,各组之间的心功能和血流动力学参数无显著差异(图S6H和S6I),而与对照组相比,增加H3K18la改善了MI后的射血分数和缩短分数,表明H3K18la除了具有抗炎和促血管生成作用外,还改善了MI后的心功能障碍(图5L至5N)。
综上所述,心肌梗死后早期单核巨噬细胞组蛋白乳酸化的增强有利于心肌梗死后的修复环境,有助于心肌梗死后心功能的改善。
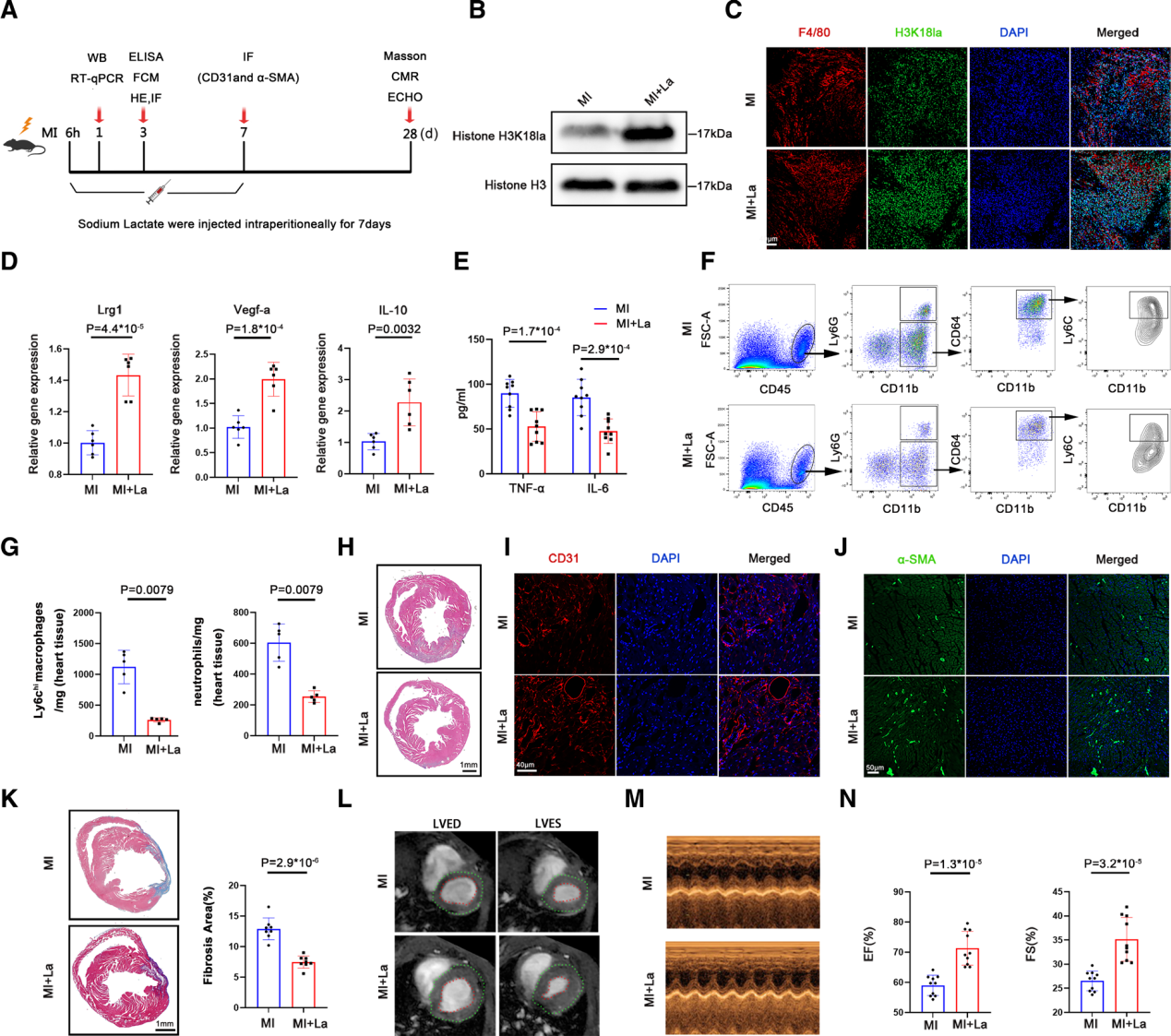
图5:H3K18la升高可改善心肌梗死后的心脏修复
(6) 抑制组蛋白乳酸化促进心肌梗死后的有害炎症和心功能障碍
图S8A显示FX-11治疗MI小鼠抑制组蛋白乳酸化的实验方案。循环单核细胞和浸润性巨噬细胞中H3K18la水平降低(图S6C、S6D、S8B、S8C),注射FX-11小鼠中H3K18la靶基因mRNA水平显著降低(图S8D)。与H3K18la升高对乳酸钠注射的影响相反,H3K18la的抑制促进了心肌梗死后血清炎症因子的表达,增加了心肌炎症细胞的浸润(图S8E至S8H),导致血管生成受阻(图S6G, S8I, S8J),病理性心脏重构加重,心功能丧失(图S6L、S6M、S8K至S8N),进一步支持组蛋白乳酸化促进内源性免疫稳态和心肌梗死后心脏修复。
(7) H3K18la操纵的单核细胞输血调节心肌梗死后的心脏修复
为探索H3K18la操纵的单核细胞经乳酸钠或FX-11处理后是否能将治疗效果转移到心肌梗死小鼠身上,从小鼠外周血中分离的循环单核细胞分别用乳酸钠(La-Mo)、FX-11(FX-Mo)或PBS (Con-Mo)刺激24小时,并用PKH26标记,在心肌梗死后6小时通过尾静脉输注到心肌梗死小鼠体内(图S9A)。这些单核细胞定位于梗死心脏(图S10A)。
与全身给药结果相似,与对照组相比,H3K18la (La-Mo)升高的单核细胞再输注显著减少炎症细胞浸润,抑制过度心肌纤维化,改善心功能,促进血管生成(图S9B至S9H, S10B)。相比之下,FX-Mo组表现出相反的效果,包括炎症增加、血管生成受限、心脏重塑不良和心功能恶化(图S9B至S9H, S10B)。
(8) 心肌梗死后循环单核细胞内源性糖酵解重编程部分促进H3K18la
巨噬细胞糖酵解通过提供乳酸作为底物来调节组蛋白乳酸化。但单核细胞在心脏募集前的代谢转变及其在心肌梗死后调节乳酸化水平中的作用尚不清楚。我们使用XF24细胞外通量分析仪测量心肌梗死后1天循环单核细胞的主要代谢参数,发现心肌梗死小鼠单核细胞的基础细胞外酸化率相对较高,糖酵解储备增加,表明心肌梗死小鼠具有较高的糖酵解代谢能力。耗氧量测量显示,心肌梗死小鼠单核细胞的基础氧化磷酸化和备用呼吸能力降低。线粒体与氟羰基氰基苯腙解偶联后,最大呼吸显著减少,表明心肌梗死后外周单核细胞更依赖糖酵解而非氧化代谢(图6A至6D)。这些数据表明单核细胞在心肌梗死后经历代谢重编程,这可能对依赖糖酵解的组蛋白乳酸化至关重要。
为验证细胞内代谢模式和细胞外代谢物对乳酸化和乳酸化相关基因表达的调节,我们使用C2HCl2NaO2(DCA,丙酮酸脱氢酶激酶抑制剂)和鱼藤酮(线粒体呼吸链复合物I的抑制剂)调节糖酵解途径和线粒体呼吸链中涉及的关键酶的活性(图6E)。用鱼藤酮治疗24小时后,BMDMs细胞内乳酸和H3K18la水平显著升高,而在乳酸脱氢酶抑制剂FX-11存在时则降低(图6F和6G)。ChIP-qPCR和RT-qPCR显示,鱼烯酮显著增加了乳酸化修饰和乳酸化靶基因Lrg1、Vegf-a和IL-10的表达(图6H和6I)。相比之下,DCA治疗后BMDMs细胞内乳酸和H3K18la水平降低。在DCA处理的细胞中再次添加乳酸成功地恢复了细胞内乳酸、组蛋白乳酸化水平和H3K18la修饰基因的表达(图6J至6M)。
为进一步验证细胞外乳酸在组蛋白乳酸化中的作用,我们使用了乳酸转运蛋白MCT1(单羧酸转运蛋白1)抑制剂AZD-3965。外源性乳酸的添加显著增加了BMDMs中组蛋白乳酸化水平和靶基因表达,而MCT1抑制显著抑制了外源性乳酸的这些作用,这表明细胞外环境中的乳酸至少部分需要通过MCT1转运到细胞内,才能参与乳酸化(图6N至6P)。

图6:代谢重编程调节单核细胞中的H3K18la
(9) IL-1β依赖性GCN5募集促进H3K18la和修复性基因表达
HAT(组蛋白乙酰转移酶) P300是潜在的组蛋白赖氨酸乳酸化写蛋白,在心肌梗死中促进单核细胞组蛋白乳酸化的机制尚不清楚。为评估三个主要HAT家族中的代表性的P300、MOF和GCN5(一般对照非抑制性5)在心肌梗死后单核细胞组蛋白乳酸化调节中的催化活性,进行了分子对接研究,以评估H3K18la与P300、MOF和GCN5的结合模式(图7A至7C)。免疫沉淀法评估P300/MOF/GCN5与乳酸化组蛋白的相互结合(图7D)。免疫荧光染色显示H3K18la和P300/MOF/GCN5在心肌梗死后1天共定位于单核细胞的细胞核(图7E)。
转染适当的小干扰(si)RNAs使HAT沉默(图S11A至S11C)。P300敲低导致BMDMs中组蛋白乳酸化水平显著降低,这与先前研究一致。GCN5敲低显著抑制H3K18la水平,ChIP-qPCR和RT-qPCR显示GCN5沉默后H3K18la修饰和H3K18la靶基因的表达水平显著降低,证实GCN5是乳化的书写者并介导了靶基因表达的调控(图7F至7J)。我们还评估了P300和MOF对H3K18la修饰和乳酸化基因转录水平的调控作用(图S11D至S11I)。
先前研究报道了IL-1β特异性募集GCN5在炎症调节中的作用。我们进一步研究IL-1β是否是心肌梗死后驱动乳酸化修饰的一个因素,IL-1β在心肌梗死后作为一种强炎性小体引发剂急剧上升。WB显示,IL-1β刺激使BMDMs中H3K18la水平呈剂量依赖性增加(图7K)。IL-1β刺激增加了GCN5和乳酸化组蛋白的相互结合(图7M),并增加了H3K18la靶基因的乳酸化和表达(图7N和7O)。为探究IL-1β与IL-1R1的结合对于IL-1β介导的GCN5依赖性乳酸化是否不可或缺,我们在IL-1β刺激下阻断了IL-1R1(图S11J)。IL-1R1阻断部分抑制了IL-1β对GCN5募集、H3K18la修饰、GCN5与乳酸化组蛋白结合以及H3K18la靶基因表达的促进作用(图7L至7O)。
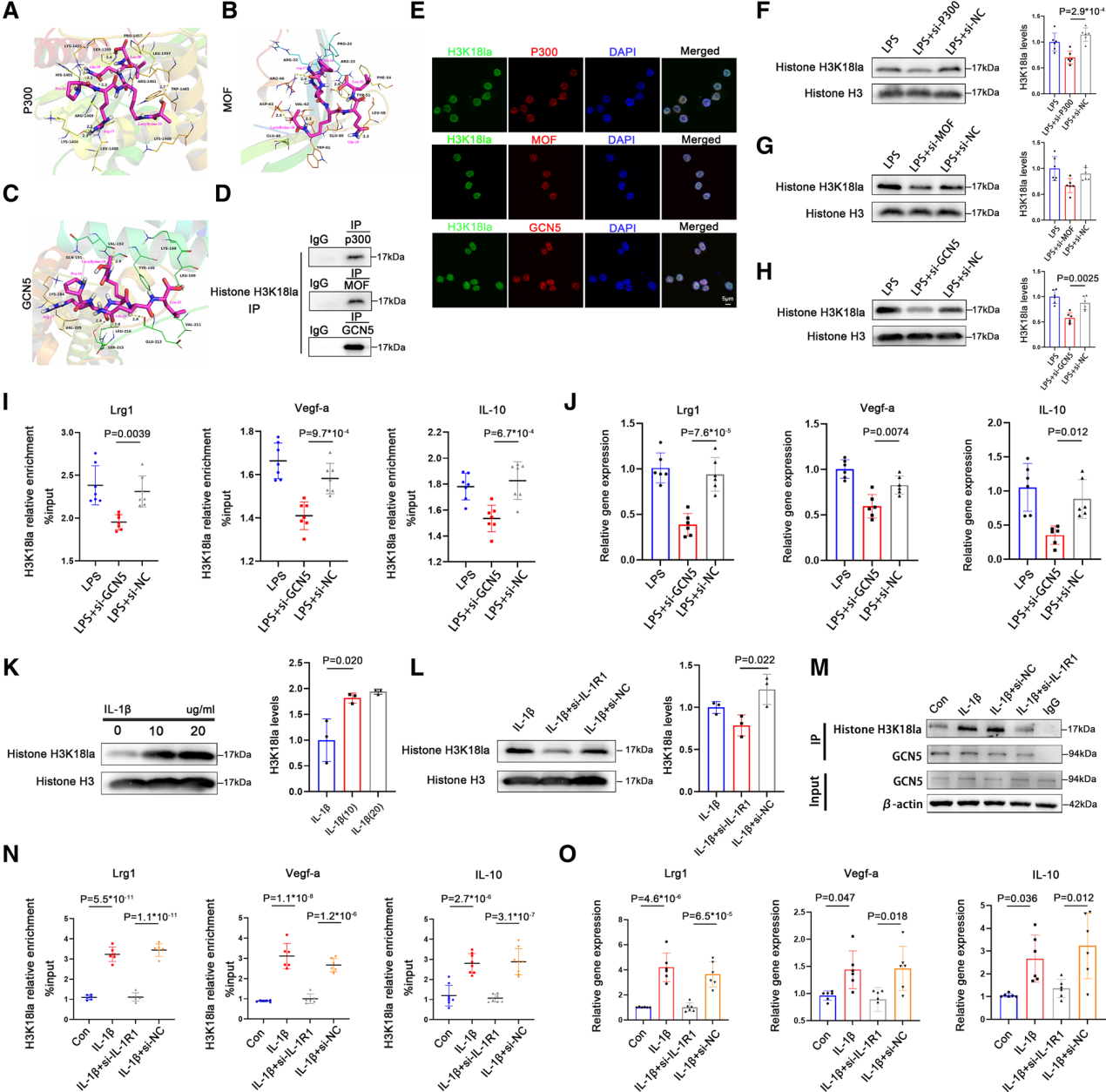
图7:IL-1β依赖性募集GCN5促进H3K18la水平和修复性基因表达
(10) 组蛋白乳酸化有利于IR损伤后心脏修复环境
为验证单核细胞H3K18la促进心脏修复的作用是否在再灌注心肌梗死中也可检测到,我们诱导了一个暂时的LAD结扎模型,然后再血运重建,以研究乳酸化在缺血再灌注(IR)损伤中的潜在作用。IR小鼠循环单核细胞的免疫印迹显示,H3K18la水平早在IR后4小时达到峰值,此后逐渐下降,直到72小时恢复到基线水平(图8A)。IR后单核细胞亚组中H3K18la水平的变化与总单核细胞基本一致(图S12A和S12B)。与永恒性LAD连接MI模型类似,IR后单核细胞Lrg1、Vegf-a和IL-10结合区域的mRNA水平和H3K18la富集显著上调(图8B和8C)。
我们在IR后6小时进行了H3K18la操纵的单核细胞再输血,观察不同处理的单核细胞的心脏定位(图8D)。与永恒性缺血类似,相对于IR损伤后的对照组,乳酸化(La-Mo)升高的单核细胞导致心肌梗死面积减小(图8E和8F),炎症细胞浸润减少(图8G),心功能改善(图8H和8I)。FX-Mo组正好相反(图8E至8I)。
综上所述,单核细胞组蛋白乳酸化在IR进展中的免疫稳态、组织修复和心功能改善中发挥积极作用。

图8:组蛋白乳酸化有利于缺血再灌注(IR)损伤后心脏修复环境
结论
组蛋白乳酸化有利于心肌梗死后单核细胞的早期远程修复基因激活,从而提高了我们对心肌梗死内源性保护机制的认识。本研究中发现的组蛋白乳酸化相关分子可能为心肌梗死后心脏修复的改善提供新的机制基础。
实验方法
RNAseq分析,WB,qRT-PCR和ChIP-qPCR,流式细胞术,EdU标记,transwell,划伤/伤口迁移和管形成试验,单核细胞再输血实验
参考文献:Wang, N., Wang, W., Wang, X., Mang, G., Chen, J., Yan, X., Tong, Z., Yang, Q., Wang, M., Chen, L., Sun, P., Yang, Y., Cui, J., Yang, M., Zhang, Y., Wang, D., Wu, J., Zhang, M., & Yu, B. (2022). Histone Lactylation Boosts Reparative Gene Activation Post-Myocardial Infarction. Circulation research, 131(11), 893–908. https://doi.org/10.1161/CIRCRESAHA.122.320488.