






CD73抑制cGAS-STING并与CD39协同促进胰腺癌
实验方法:MetaGx胰腺基因表达数据集采集,基因签名,定量免疫荧光分析,ELISAs,Western blots,CD39 IHC分析,TILs的FACS分析,实时定量PCR,球体形成和OT-I活化试验,体外增殖试验,g-H2AX免疫荧光
外核苷酶CD39和CD73催化细胞外ATP产生免疫抑制腺苷,代表了潜在的癌症靶点。我们通过临床样本和实验小鼠肿瘤研究CD39和CD73在胰腺导管腺癌(PDAC)中的生物学影响。在人类PDAC样本中,基质CD39和肿瘤CD73的表达与较差的生存率显著相关,并消除了与肿瘤浸润CD8+ T细胞存在相关的有利预后影响。在小鼠移植的KPC肿瘤中,骨髓细胞上的CD39和CD73以及肿瘤细胞上的CD73均可促进浸润性骨髓细胞向M2样表型极化,从而促进肿瘤生长。肿瘤特异性CD8+ T细胞和胰腺星状细胞上的CD39也抑制T细胞产生IFNg。虽然治疗性抑制CD39或CD73在体内显著延缓肿瘤生长,但靶向这两种外核苷酶显示出明显更好的抗肿瘤活性。CD73在人和小鼠PDAC肿瘤细胞上的表达也对吉西他滨和辐照诱导的DNA损伤具有保护作用。人类PDAC细胞系的大规模药物基因组学分析揭示了CD73表达与吉西他滨化疗耐药之间的显著关联。CD73缺陷肿瘤细胞中DNA损伤的增加与cGAS-STING通路的激活有关。CD73抑制剂AB680的体内抗肿瘤活性需要cGAS在小鼠KPC肿瘤细胞中的表达。我们的研究阐明了CD73和CD39似乎合作促进PDAC进展的分子机制。
技术路线:

结果:
(1) 肿瘤CD73和间质CD39都与PDAC的低生存率相关
为研究PDAC中腺苷通路对预后的影响,我们在12项独立研究中对1200多名PDAC患者进行了CD73和CD39的基因表达荟萃分析。CD73与较差的OS显著相关(图1A)。CD39总体上与较好的OS相关,尽管这在单个队列中没有达到统计学意义(补充图未展示)。我们在两个最大的队列中探讨了CD73和CD39之间的预后关联。只有当PDAC肿瘤同时具有高CD39表达时,才观察到CD73与较差预后的关联(图1B)。两个最大队列的相关分析进一步揭示了CD39基因表达与细胞毒性T细胞、Tregs和T细胞衰竭标志物(即PDCD1、CTLA4、TIGIT、LAG3)之间的显著正相关,而CD73则没有显著相关性(图1C)。
我们研究了来自独立队列的PDAC手术样本的上皮和间质中CD73(图2A)和CD39(图2B)蛋白表达的组织学分布。与匹配的正常邻近胰腺组织相比,CD73在PDAC肿瘤上皮中显著上调,而在肿瘤间质中表达微弱但显著下调(图2C)。CD39在PDAC肿瘤间质中显著上调(图2D)。肿瘤上皮CD73与高分期和低分化PDAC肿瘤之间存在显著关联。而CD39与临床病理特征之间无相关性。
生存分析显示,更高的肿瘤上皮CD73表达(图2E)和更高的肿瘤间质CD39表达(图2F)与PDAC患者更差的OS相关。在多变量分析中,当调整术前CA 19-9、淋巴结状态、分化和切除边缘时,肿瘤上皮CD73表达与较差的OS相关。结合上皮CD73和间质CD39表达的生存分析提供了进一步的预后(补充图未展示)。

图1:CD73基因表达与PDAC预后不良相关
(2) CD73和CD39消除了肿瘤浸润性CD8+ T细胞的良好预后影响
正如在其他PDAC队列中所显示的那样,肿瘤浸润性CD8+ T细胞的存在与OS改善相关(补充图未展示)。支持我们的基因表达数据(图1C),CD39蛋白表达与CD8+ T细胞浸润呈正相关,而CD73蛋白表达与肿瘤浸润的CD8+ T细胞不相关(补充图未展示)。我们研究了CD73或CD39表达是否影响PDAC免疫监测。肿瘤上皮CD73(图2G)或肿瘤间质CD39(图2H)的高表达完全取消了肿瘤浸润性CD8+ T细胞在PDAC患者中的良好预后价值。

图2:肿瘤CD73和间质CD39蛋白表达与PDAC预后不良和免疫监视抑制相关
(3) 可溶性CD73与更差的PDAC生存相关
由于CD73可以从细胞表面切割并以可溶性形式存在,我们评估了可溶性CD73 (sCD73)是否可以作为PDAC的无创预后生物标志物。在一项独立队列研究中,良性或恶性胰腺肿瘤患者的sCD73水平明显高于健康供体(图2I)。PDAC患者较高的sCD73与较高的CA 19-9水平、位于胰腺头部的肿瘤、较高的分期和淋巴血管侵犯相关(补充表未展示)。较高的sCD73也与较差的OS显著相关(图2J)。然而,在多变量分析中,sCD73没有达到显著性。我们评估了血清中sCD73是否可以作为PDAC肿瘤中CD73表达的替代指标。在亚队列中,未观察到可溶性和肿瘤CD73水平之间的相关性。
(4) 肿瘤细胞和髓样细胞上的CD73有利于小鼠PDAC中M2样髓样细胞的浸润
我们利用Crispr/Cas9介导的基因缺失研究了肿瘤来源的CD73在小鼠KPC或人PANC1肿瘤细胞系中的作用。KPC和PANC1表达CD39为阴性(补充图未展示)。在体外,肿瘤来源的CD73对肿瘤细胞增殖无影响(补充图未展示)。肿瘤源性CD73的缺失显著降低了KPC肿瘤在体内的生长,并改善了吉西他滨活性(图3A)。这与肿瘤IFNg水平的显著增加以及IL10和精氨酸酶-1的微弱但显著的增加相关(图3B)。肿瘤源性CD73的缺失还显著降低了KPC肿瘤中M2样肿瘤相关巨噬细胞(TAM)和单核髓源性抑制细胞(M-MDSC)的浸润和极化(图3C)。靶向肿瘤来源的CD73对CD8、CD4或粒细胞(G)-MDSC浸润没有影响。由于CD73在KPC浸润的髓细胞上表达,我们评估了髓源性CD73的作用。条件CD73fl/fl小鼠与LysMCre+/−小鼠(骨髓特异性CD73缺失),并用CD73阴性的KPC肿瘤细胞攻击。髓源性CD73显著促进KPC肿瘤生长(图3D),促进M2样TAMs的浸润(图3E)和M2极化。骨髓来源的CD73对肿瘤浸润性CD8、CD4、M-MDSCs或G-MDSCs没有影响(补充图未展示)。
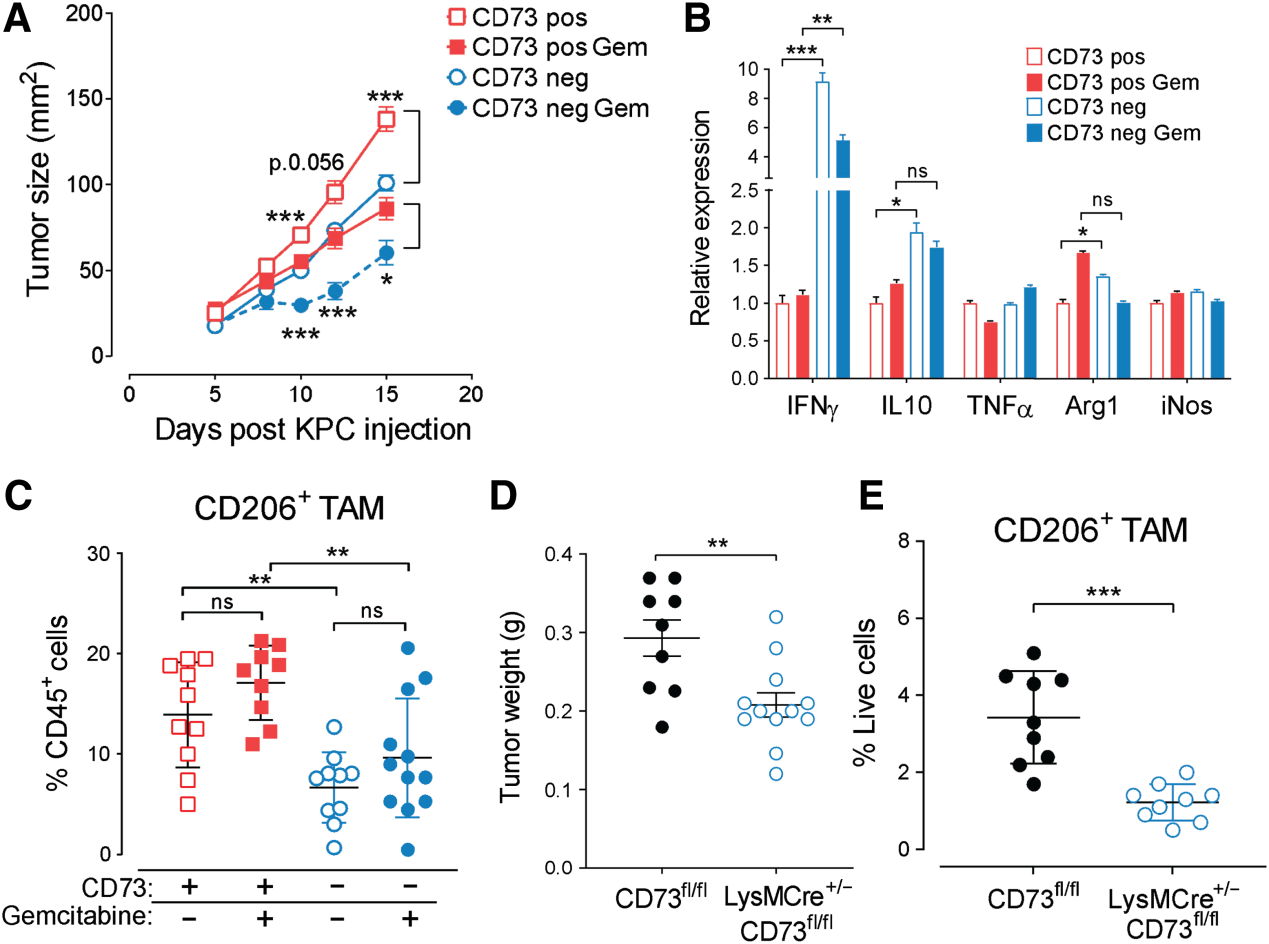
图3:肿瘤细胞和骨髓细胞上的CD73促进小鼠PDAC
(5) 骨髓细胞、成纤维细胞和T细胞上的CD39抑制抗PDAC免疫反应
我们研究了CD39的作用。由于髓系细胞是KPC肿瘤中CD39的主要来源,我们评估了在条状CD39fl/fl LysMCre+/−用KPC肿瘤细胞攻击小鼠。髓系细胞CD39缺失显著降低KPC肿瘤生长(图4A)和显著减少M2样TAM浸润(图4B)。与我们在CD73中观察到的情况相反,骨髓特异性缺失CD39还与肿瘤浸润性CD8+ T细胞和CD4+Foxp3− T细胞(图4C)。
我们评估了CD39在肿瘤相关成纤维细胞和效应CD8+ T细胞中的作用。我们使用体外球体培养物来测量T细胞的功能,球体培养物由表达OVA的KPC肿瘤细胞、原代胰腺星状细胞(WT或CD39−/−)和OVA特异性CD8+ OT-I细胞(WT或CD39−/−;图4D)。CD39−/− OT-I CD8+ T细胞,以及CD39−/−胰腺星状成纤维细胞,每一种都显著增加了OT-I效应T细胞产生的IFNγ (图4D)。

图4:CD39与CD73协同促进小鼠PDAC
(6) 靶向CD39和CD73增强吉西他滨抗小鼠PDAC的活性
我们评估了CD39是否代表针对PDAC的治疗靶点。从第7天开始,用阻断性单克隆抗小鼠CD39治疗已建立的KPC肿瘤(>25 mm2),每周3次,单独或联合吉西他滨(100 mg/kg,第7天和第10天)。抗CD39单药治疗显著延缓了KPC肿瘤的生长,并显著增强了吉西他滨对已建立肿瘤的活性(图4E)。同样,使用选择性CD73抑制剂(即AB680)治疗也显著抑制了KPC肿瘤的生长,增强了吉西他滨的活性(图4F)。
我们评估CD39和CD73是否具有冗余的致瘤作用。将KPC肿瘤注射到WT和CD39−/−用CD73抑制剂AB680联合吉西他滨治疗。与吉西他滨联合靶向宿主来源的CD39进一步增强了CD73抑制的治疗活性(图4G)。使用不同的KPC模型(即KPC 1199细胞)。用AB680、单克隆抗CD39或两者同时治疗的KPC-OVA肿瘤的免疫分析显示,只有联合抑制CD73和CD39才能显著增加肿瘤特异性T细胞浸润,并阻止PD-1衰竭标志物的获得(图4H)。我们的结果揭示了CD73和CD39在促进PDAC中的非冗余作用。
(7) 肿瘤来源的CD73保护DNA损伤并抑制cGAS-STING
由于PDAC细胞表达高CD73,但不表达CD39,我们研究了CD73是否以细胞自主的方式赋予PDAC肿瘤细胞增殖优势。CD73基因沉默在正常培养条件下对小鼠或人PDAC细胞的存活和增殖没有影响。然而,在KPC或PANC1细胞中,CD73缺失显著增强了肿瘤细胞对吉西他滨的体外敏感性(图5A, B)。为了进一步评估CD73在PDAC化疗耐药中的作用,我们在三个最大的药物基因组学药物筛选研究中确定CD73基因表达是否与吉西他滨敏感性相关。PDAC细胞系中CD73基因表达与吉西他滨反应呈显著负相关(C-index=0.62, P<0.001;图5C),由药物剂量-反应曲线上方的面积测量。与之一致的是,gH2AX染色显示,吉西他滨治疗CD73缺陷的KPC肿瘤细胞诱导了更大的DNA损伤(图5D, E)。CD73表达的部分恢复恢复了表型并减少了DNA损伤(补充图未展示)。在辐照反应(图5F)和用CD73抑制剂AB680处理的人PANC1肿瘤细胞(图5G)中获得了类似的结果。用选择性激动剂(BAY 60-6583)激活A2B受体恢复了这种表型,正如吉西他滨处理的PANC1在A2B受体激活(图5G)和PKC细胞(图5H)后DNA损伤显著减少所揭示的那样。
DNA传感器cGAS可以通过产生cGAMP将DNA损伤与炎症联系起来,cGAMP反过来激活STING依赖基因转录。由于CD73表达可以保护PDAC细胞免受DNA损伤,我们研究了CD73是否可以抑制cGAS-STING的激活。与吉西他滨治疗后CD73缺失的肿瘤细胞相比,CD73缺失的KPC细胞产生了显著更高的cGAMP(图5I),表达了显著更高的STING靶基因(图5J)。这提示cGAS-STING通路可能在CD73抑制的治疗活性中起作用。为了验证这一点,我们通过Crispr/Cas9从亲本KPC细胞中删除了cGAS,生成了一个由确认cGAS-STING激活缺失的单个克隆组成的多克隆群体,并评估了AB680对cGAS缺失的KPC肿瘤和控制表达Cas9的KPC肿瘤的治疗活性。肿瘤细胞中cGAS的缺失消除了AB680的体内活性(图5K)。使用基于shRNA的cGAS敲除也获得了类似的结果(补充图未展示)。我们的数据突出了CD73在调节cGAS-STING通路中的作用,并强调了肿瘤cGAS激活对CD73抑制剂治疗活性的重要性。
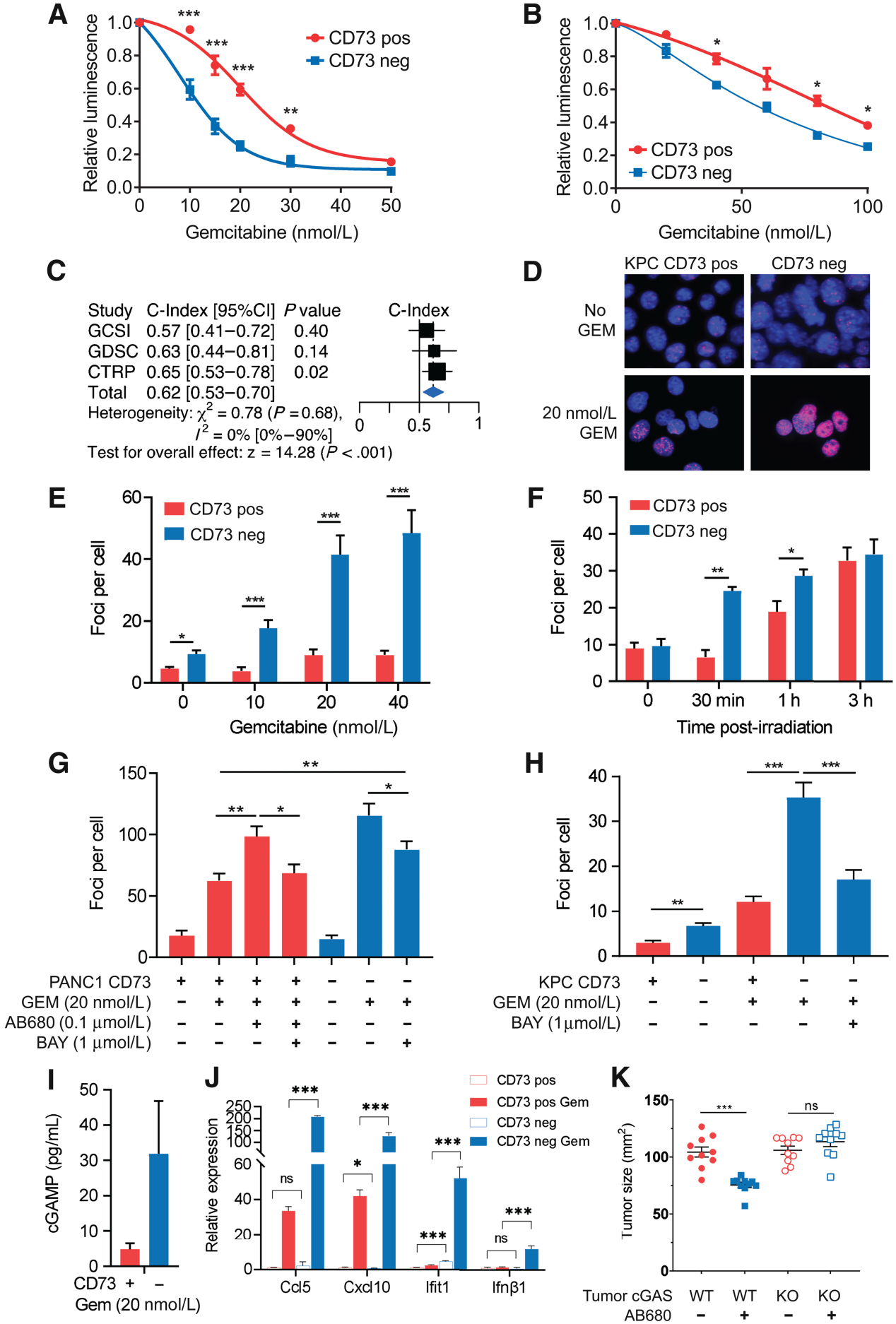
图5:CD73促进DNA损伤修复并抑制cGAS-STING激活
结论:我们的研究支持进一步评估CD39和CD73在PDAC中的生物学作用,作为免疫检查点和对吉西他滨产生化学耐药。我们证明CD39和CD73都与不良的临床结果显著相关,并抑制肿瘤对PDAC的免疫监视。我们建议靶向CD39-CD73轴将有利于免疫冷肿瘤,如PDAC。
参考文献:Jacoberger-Foissac, C., Cousineau, I., Bareche, Y., Allard, D., Chrobak, P., Allard, B., Pommey, S., Messaoudi, N., McNicoll, Y., Soucy, G., Koseoglu, S., Masia, R., Lake, A. C., Seo, H., Eeles, C. B., Rohatgi, N., Robson, S. C., Turcotte, S., Haibe-Kains, B., & Stagg, J. (2023). CD73 Inhibits cGAS-STING and Cooperates with CD39 to Promote Pancreatic Cancer. Cancer immunology research, 11(1), 56–71. https://doi.org/10.1158/2326-6066.CIR-22-0260.