






FLT4/VEGFR3激活AMPK,以协调糖代谢重编程与自噬和炎症小体激活
巨噬细胞快速进行糖酵解重编程响应巨噬/自噬,炎症小体激活和清除细菌。识别参与其中的关键分子将提供关键的潜在治疗应用。作者在本研究中阐明了LT4-AMPK模块协调糖酵解重编程、自噬、炎症小体激活和焦死来消灭入侵细菌的分子机制。本研究2021年10月发表在《AUTOPHAGY》,IF=9.77。
技术路线:

主要结果:
1. FLT4/VEGFR3激活AMPK,以协调糖代谢重编程与自噬和炎症小体激活,以消除细菌
为了了解糖酵解重编程的改变是如何与细菌清除联系在一起的,作者在flt4WT/WT(野生型)小鼠细胞内感染来源于鼠伤寒菌株SL1344菌株后,检测了靶器官肝脏中的糖代谢谱和伤寒链球菌的数量。结果发现具有直接抗菌活性的柠檬酸和NAD (P) H在肝脏感染后大量增加。相比之下,与PBS处理的flt4WT/WT小鼠相比,SL1344感染的flt4WT/WT小鼠肝脏中琥珀酸盐和乳酸盐的浓度降低,可促进促炎巨噬细胞功能(Figure 1A)。因为之前证明了flt4突变小鼠缺乏细胞外配体结合域(LBD:缺乏免疫球蛋白样域2和3,命名为flt4ΔLBD/ΔLBD)更容易感染。因此,作者比较了Flt4WT/WT小鼠和flt4ΔLBD/ΔLBD小鼠的糖酵解重编程变化。与Flt4WT/WT小鼠不同, flt4ΔLBD/ΔLBD小鼠柠檬酸和NAD(P)H未能增加。此外,Flt4WT/WT小鼠琥珀酸和乳酸的产量减少,而突变小鼠显示出这些代谢物的产量增加(Figure 1B-F)。此外,flt4ΔLBD/ΔLBD小鼠肝脏和脾脏中鼠鼠伤寒杆菌数量显著增加(Figure 1G)。
作者通过评估巨噬细胞内鼠伤寒链球菌的菌落形成单位(cfu)来研究FLT4如何影响细菌清除。与体内抗细菌反应缺陷相似,flt4ΔLBD/ΔLBD巨噬细胞与Flt4WT/WT巨噬细胞相比,携带的鼠伤寒杆菌数量要多得多。 由于包括巨噬细胞在内的吞噬细胞可以吞噬固体颗粒,这是杀死细菌的关键过程,作者首先检测了FLT4信号通路是否影响吞噬能力。将GFP(绿色荧光蛋白)-大肠杆菌(e.c oli-GFP)与flt4ΔLBD/ΔLBD小鼠或与flt4WT/WT同窝的小鼠的腹腔渗出物巨噬细胞PEM(腹腔渗出物巨噬细胞)共孵育,流式细胞术检测结果显示flt4ΔLBD/ΔLBD PEMs对大肠杆菌GFP的吞噬量低于Flt4WT/WT PEMs (Figure 1I)。溶酶体的酶可以帮助巨噬细胞消灭细菌。接下来,作者评估了吞噬酶体的成熟是否受到flt4的调控。聚焦显微镜成像显示内化的鼠伤寒杆菌GFP与装载LysoTracker Red的野生型BMDMs共定位(Figure 1J)。综上所述,作者的数据表明,flt4可以通过增加吞噬、ROS的产生和自噬小体的成熟来促进细菌的清除,并伴随着糖代谢的改变。
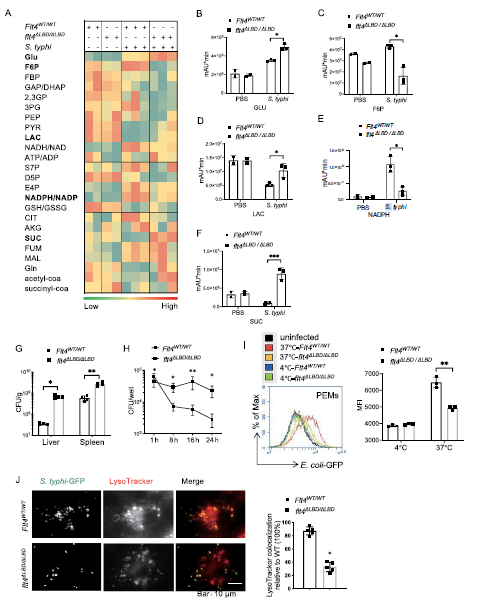
Fig 1 巨噬细胞表面受体flt 4控制糖酵解重编程和细菌清除
2. FLT4在清除鼠伤寒杆菌过程中保护巨噬细胞免遭细胞凋亡
鼠伤寒链球菌感染可导致巨噬细胞凋亡,其特征是细胞肿胀,膜囊迅速扩张、破裂和核凝结。在图2A中,在伤寒沙门氏菌感染期间,flt 4ΔLBD/ΔLBDPEMs细胞死亡数量增加,呈肿胀形状(Figure 2B),在FLT4WT/WT巨噬细胞中,存在大量含有细菌残体的吞噬-溶酶体空泡。感染的flt4ΔLBD/ΔLBD巨噬细胞的细胞死亡率高于Flt4WT/WT细胞(Figure 2C, D)。由于甘氨酸处理可降低胞质释放水平乳酸脱氢酶(LDH)和保护细胞免于焦亡,作者用这个方法来鉴定焦亡。作者发现甘氨酸治疗可以保护flt4ΔLBD/ΔLBD或Flt4WT/TKmut的巨噬细胞,其保护水平与WT细胞在伤寒沙门氏菌感染后的保护水平相当(Figure 2E)。总之,这些数据表明,flt4保护巨噬细胞免受SL-1344感染引起的细胞凋亡。
炎症小体的激活和随后的焦亡是抵抗细菌感染的关键防御。因此,作者通过ELISA分析细胞上清液中成熟IL1B浓度来研究炎症小体的活化。事实上,FLT4ΔLBD/ΔLBD PEMs或BMDMs分泌的IL1B水平远高于对照细胞(Figure 2F, G)。此外,flt4ΔLBD/ΔLBD巨噬细胞中IL1B的增加与感染FLT4ΔLBD/ΔLBD小鼠的葡萄糖代谢产物包括琥珀酸和乳酸盐的增加是一致的(Figure 1A),这两者都被认为是促进炎症的。已有研究报道CASP1和GSDMD (蛋白D)诱导细胞焦亡,这可能对许多细菌有保护作用。然而,体内炎症小体信号的过度激活和焦亡可能对宿主有害。在作者的研究中,作者观察到IL1B浓度越高,细胞焦亡率越高,而细菌数量越多。
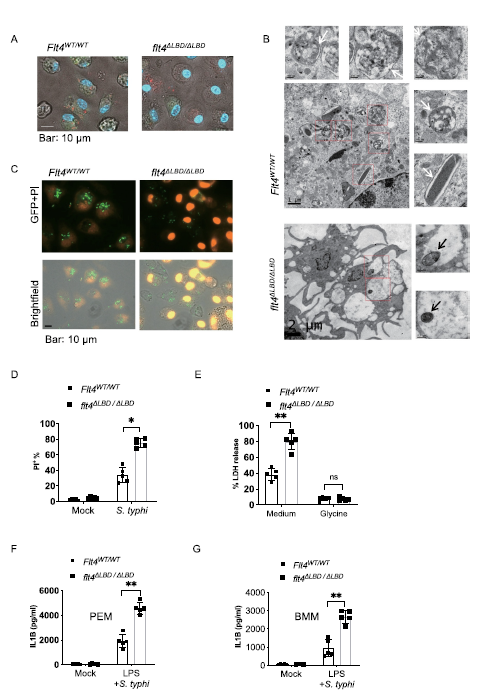
Fig 2 FLT4在清除鼠伤寒杆菌过程中保护巨噬细胞免遭细胞凋亡。
3. FLT4抑制炎症小体和CASP1的激活鼠鼠伤寒杆菌感染巨噬细胞
在炎性小体组装过程中,成熟IL1B的释放需要CASP1激活来切割前体pro-IL1B。炎性小体激活的另一个信号是PYCARD/ASC (PYD和CARD结构域包含/凋亡相关斑点样蛋白包含CARD)斑点的形成,它是CASP1激活的平台。作者发现在flt4ΔLBD/ΔLBD巨噬细胞中形成了较高比例的PYCARD斑点(Figure 3A)。与伤寒沙门氏菌感染的Flt4WT/ WT相比,检测对照细胞的蛋白质齐聚状态(Figure 3B)。结果表明,FLT4信号通路抑制细菌感染后巨噬细胞PYCARD斑点的形成。
接下来,作者测量了FLT4如何影响CASP1的激活。CASP1是一种关键酶,它能切割前il1b /白介素-1β蛋白转化为活性IL1B。CASP1与炎性小体一起能诱导其自催化活性和自裂解,从而产生催化活性亚基CASP1 / p10及CASP1 / p20。与对照组细胞相比,感染沙门氏菌之后, flt4ΔLBD/ΔLBD巨噬细胞显示出CASP1以CASP1/p20裂解表达形式增加(Figure 3C)。同样,感染沙门氏菌之后, 激酶死亡突变体Flt4WT/TKmut或FLT4的敲除显著增强了CASP1/p10的裂解形式(Figure 3D, E)。这些数据表明,FLT4信号通路抑制炎性小体的激活。
LPS启动伤寒沙门氏菌感染迅速诱导VEGFC蛋白表达(Figure 3F)。而外源性VEGFC可以降低Flt4WT/WT巨噬细胞中CASP1的激活(Figure 3G)。而FLT4抑制剂则增强了Flt4WT/WT巨噬细胞中CASP1的活化(Figure 3H)。使用外源性VEGFC和FLT4抑制剂的治疗并没有进一步影响flt4ΔLBD/ΔLBD巨噬细胞中CASP1的激活(Figure 3G, H)。这些结果说明在伤寒链球菌感染的巨噬细胞中,FLT4信号通路的激活抑制了炎症小体触发的PYCARD斑点的形成和CASP1激活。
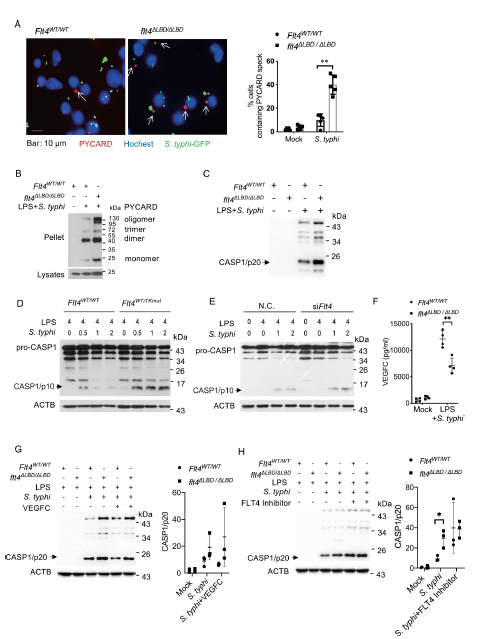
Fig 3 FLT4抑制鼠伤寒杆菌感染巨噬细胞的炎症小体和CASP1的激活
4. FLT4以自噬依赖的方式增强细菌消除和防止炎性小体激活
在巨噬细胞识别病原体或吞噬细胞质细菌的过程中,MAP1LC3以胞质形式与磷脂酰乙醇胺结合形成MAP1LC3-磷脂酰乙醇胺结合物(MAP1LC3 -II),它被招募到吞噬细胞膜上。成熟后,自噬体与溶酶体融合,降解和清除入侵的病原体,这也被称为自噬体。作者通过电子显微镜发现细菌降解野生型巨噬细胞显示边缘不规则 (Figure 4A)。
自噬酶体形成的过程涉及到吞噬体和自噬机制的组成部分。因此作者用免疫印迹法测量了鼠伤寒杆菌感染野生型或flt4∆LBD/∆LBD巨噬细胞中的MAP1LC3-I和MAP1LC3-II水平。结果发现,寒沙门氏菌感染提高了野生型巨噬细胞中MAP1LC3-II:MAP1LC3-I的比值,而flt4∆LBD/∆LBD巨噬细胞中MAP1LC3-II水平显著降低(Figure 4B)。 在永生化小鼠骨髓来源的巨噬细胞(iBMDM)细胞中过表达FLT4或外源性VEGFC处理均提高了MAP1LC3-II水平(Figure 4C, D)。此外,用FLT4抑制剂或自噬药物抑制剂3-甲基腺嘌呤(3-Ma)处理降低了MAP1LC3-II水平(Figure 4E, F)。这些发现表明,FLT4信号通路以map1lc3依赖的方式增强了自溶酶体的形成。与Flt4 WT/WT 巨噬细胞相比,flt4∆LBD/∆LBD巨噬细胞中MAP1LC3点下降,与免疫印迹结果一致。此外,flt4∆LBD/∆LBD BMDMs显示出LAMP1与MAP1LC3共定位的百分比下降。这些数据表明,FLT可以促进吞噬-溶酶体的成熟(Figure 4G)。为进一步确定FLT4信号通路是否增强自噬溶酶体的形成,通过巴弗洛霉素a1处理Flt4WT/WT和Flt4∆LBD/∆LBD巨噬细胞来阻断自噬溶酶体融合,然后转染。结果发现,与flt4WT/WT巨噬细胞相比,flt4∆LBD/∆LBD巨噬细胞中SQSTM1/p62 蛋白水平表达量增加。这表明FLT4参与了自噬溶酶体形成的调控(Figure 4H)。
由于作者观察到寒链球菌感染后,FLT4同时影响巨噬细胞的自噬和炎症小体激活,作者进一步探索自噬是否可以调节炎症小体激活。作者使用自噬抑制剂3-MA治疗伤寒链球菌感染的巨噬细胞,结果发现寒链球菌在Flt4WT/WT巨噬细胞中的清除水平与在Flt4∆LBD/∆LBD巨噬细胞中的清除水平相似(Figure 4I)。此外,3-MA治疗显著增加了成熟IL1B的分泌(Figure 4J),并且增加了CASP的裂解(Figure 4k)。此外,野生型和flt4∆LBD/∆LBD感染巨噬细胞后,3-MA处理可产生相当数量的胞内寒链球菌,同时也消除了成熟IL1B分泌、PYCARD寡聚和CASP1激活(Figure 4J-K)。总的来说,作者已经阐明了FLT4以自噬机制依赖的方式增强了细菌的消除并减弱了过度的炎症小体激活。
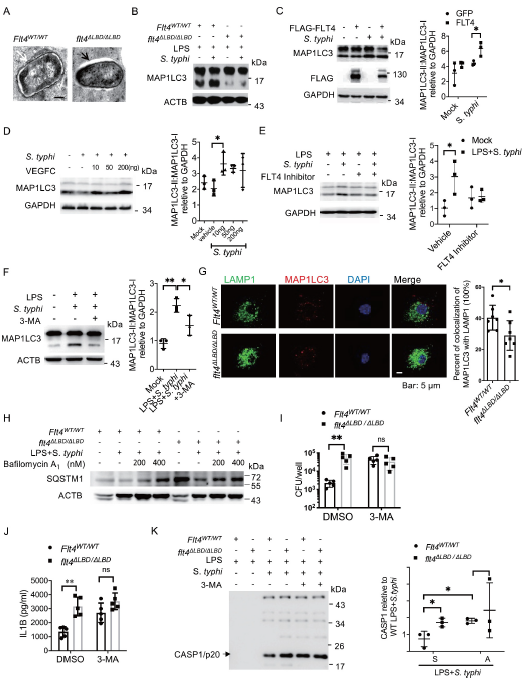
Fig 4 FLT4以自噬依赖的方式增强细菌消除和防止炎性小体激活
5. FLT4与AMPK相互作用以协调糖代谢重编程、自噬和炎症小体激活
FLT4是细胞表面的一种酪氨酸激酶受体,研究其酪氨酸激酶活性如何对其靶向下游效应物调节自噬和炎症小体激活的能力至关重要。为了识别新的结合部分,作者在RAW264.7细胞系中过表达FLT4,并进行质谱分析。作者根据它们潜在的酪氨酸修饰位点和与自噬的功能相关性确定了11个潜在的靶蛋白。采用免疫沉淀法确定FLT4和这些候选基因之间的相互作用,其中RKAA1 / AMPKα1(蛋白激酶,
amp激活,α 1催化亚基)与HEK293细胞中的FLT4相互免疫沉淀(Figure 5A)。此外,在RAW264.7细胞中,FLT4与PRKAA1对伤寒沙门氏菌感染的反应增强(Figure 5B)。沙门氏菌感染增强了PRKAA1磷酸化(p-PRKAA1)水平和MAP1LC3脂化(MAP1LC3-II)水平,这种效果通过Flt4抑制剂的治疗显著降低(Figure 5C)。相比之下,FLT4抑制剂未能进一步减弱FLT4∆LBD/∆LBD巨噬细胞的这些效应(Figure 5C)。
作者接下来评估了FLT4是否通过AMPK减少自噬。感染沙门氏菌后,flt4∆LBD/∆LBD巨噬细胞中p-PRKAA1、p-ULK1(Ser555)和MAP1LC3 脂化作用降低 (Figure 5D)。有趣的是,药物AMPK激活剂AICAR(5-氨基酰亚胺-唑-4-carboxamide核糖核苷)治疗显著提高了flt4∆LBD/∆LBD巨噬细胞的p-ULK1、p-PRKAA1和MAP1LC3脂化水平,达到了与flt4 WT/WT巨噬细胞相似的程度(Figure 5D)。这表明激活的AMPK是FLT4信号通路调控自噬的下游效应因子。此外,敲低PRKAA1降低ULK1水平和UMAP1LC3脂化水平,以及flt4WT/WT和Flt4∆LBD/∆LBD巨噬细胞的MAP1LC3脂化程度,与感染后的Flt4 WT/WT和Flt4∆LBD/∆LBD巨噬细胞相似 (Figure 5E)。此外,使用AMPK激动剂AICAR显著抑制成熟IL1B的产生,这表明AMPK抑制炎性小体激活(Figure 5F)。同样的,AICAR 处理后,成熟Flt4WT/WT和Flt4∆LBD/∆LBD巨噬细胞中IL1B和胞内细胞内沙门氏菌感染情况接近(Figure 5F,G),这些数据表明AMPK是FLT4在炎症小体激活和细胞内细菌自噬消除中的下游靶点。
由于作者观察到AMPK激动剂可以逆转flt4∆LBD/∆LBD巨噬细胞在调节自噬和炎症小体激活中的表型,接下来进一步探讨FLT4是否通过下游AMPK调控葡萄糖代谢。作者在感染时通过AMPK激活了flt4 WT/WT和flt4∆LBD/∆LBD巨噬细胞中的AMPK(Figure 5H),静息状态下,flt4WT/WT和flt4∆LBD/∆LBD巨噬细胞显示出相似的葡萄糖代谢产物。细菌感染后,与flt4WT/WT巨噬细胞相比,Flt4∆LBD/∆LBD巨噬细胞显示NADH/NAD 和 NADPH/NADP 水平显著降低。但提高乳酸和琥珀酸水平。这些数据共同表明,FLT4和AMPK在巨噬细胞中共同作用,重新编程糖酵解代谢,这与炎症小体激活和自噬有关。
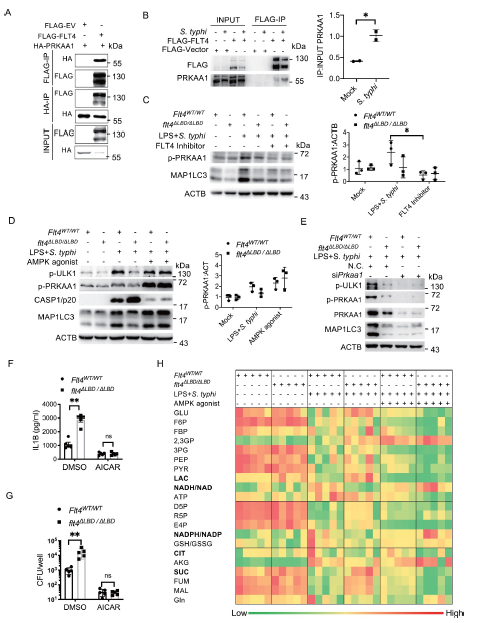
FIG 5.FLT4与AMPK相互作用以协调糖代谢重编程、自噬和炎症小体激活
6. flt4诱导的PRKAA1酪氨酸磷酸化对自噬和炎症小体的激活是必不可少的
接下来,作者研究了PRKAA1中的哪些酪氨酸可以被酪氨酸激酶受体FLT4磷酸化。FLT4过表达促进了PRKAA1酪氨酸磷酸化(Figure 6A)。根据UniPort数据库的质谱分析结果,作者确定了PRKAA1中5个可能的酪氨酸磷酸化位点,包括Y247 (A1)、Y294 (A2)、Y441/442 (A3)和Y500 (A5),作者在每个位点进行了了苯丙氨酸点突变(Fig 6B),并将它们分别转染到Hela细胞。与野生型对照(A0)相比,A1和A3突变可显著降低MAP1LC3脂化(Fig 6C,D)。作者进一步的通过PRKAA1突变过表达FLT4,发现A1和A3突变也显著阻断FLT4诱导MAP1LC3脂化(Fig 6D)。这些数据表明,Y247和Y441/442的磷酸化在PRKAA1在自噬中起着至关重要的作用。
为进一步确定PRKAA1酪氨酸在Y247和Y441/442位点磷酸化未知的功能,作者将野生型PRKAA1的 A1、A3突变稳定表达到iBMDM中(Figure 6E)。在鼠伤寒杆菌感染后,A0 iBMDMs中PRKAA1和ULK1的磷酸化增加,而 A1或A3突变体的p-PRKAA1和p-ULK1水平显著降低(Figure 6E)。与此一致的是,与GFP对照相比,PRKAA1过表达而非A1和A3突变体增强了细菌清除率(Figure 6G),
为了确定PRKAA1是否也能调节炎症小体的激活,作者用鼠伤寒杆菌感染了表达PRKAA1或A1或A3突变体的iBMDM。作者发现PRKAA1过表达减少了成熟巨噬细胞产生IL1B,但A3突变体(Y441/442 F)未显示抑制作用(Figure 6H).
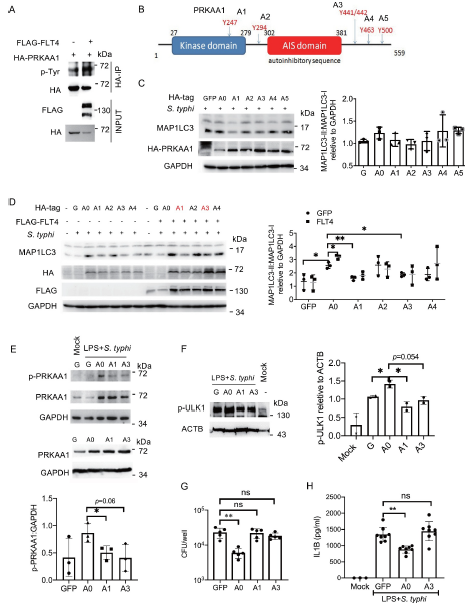
FIG 6 flt4诱导的PRKAA1酪氨酸磷酸化对自噬和炎症小体的激活是必不可少的
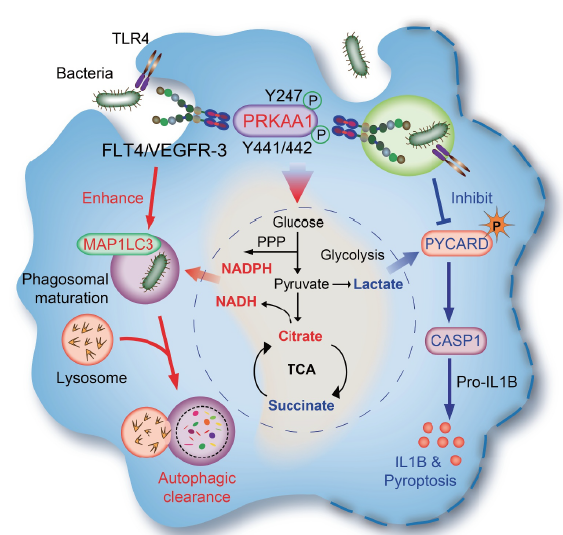
FIG 7 flt4-AMPK调节巨噬细胞糖酵解代谢,以协调细菌感染期间的自噬、焦亡和炎症小体激活。
综上所述,作者的首次发现提出flt 4磷酸化PRKAA1中的Y247和Y441/442对糖酵解重编程、细菌清除和炎症小体激活至关重要(Figure 7)。
参考文献:
Ma, L., et al., FLT4/VEGFR3 activates AMPK to coordinate glycometabolic reprogramming with autophagy and inflammasome activation for bacterial elimination. Autophagy, 2021: p. 1-16.