






p53/microRNA-214/ULK1轴—糖尿病肾病治疗的靶点
糖尿病肾病(DKD)中自噬调节失调已有报道,但其潜在机制及其致病作用仍不清楚。今天小编为大家介绍近期发表于影响因子11.864的杂志“The Journal of Clinical Investigation”的文献“p53/microRNA-214/ULK1 axis impairs renal tubular autophagy in diabetic kidney disease”,带大家了解自噬与DKD发展的机制。

在本文中,自噬在DKD模型和人类糖尿病肾脏中受到抑制。DKD的自噬损伤与ULK1的下调有关,ULK1由糖尿病肾细胞和组织中miR-214的上调介导。从肾脏近端小管切除miR-214可防止糖尿病肾脏ULK1表达降低和自噬受损,从而减少肾脏肥大和蛋白尿。此外,p53的阻断减弱了miR-214在DKD的诱导,导致更高水平的ULK1和自噬,伴随着DKD的改善。与非糖尿病样本相比,糖尿病患者的肾活检显示p53和miR-214的诱导,与ULK1和自噬的下调有关。我们还发现p53/miR-214与肾纤维化呈正相关,但ULK1/LC3与糖尿病患者的肾纤维化呈负相关。
技术路线:
结果:
1) 糖尿病小鼠肾小管细胞自噬减少
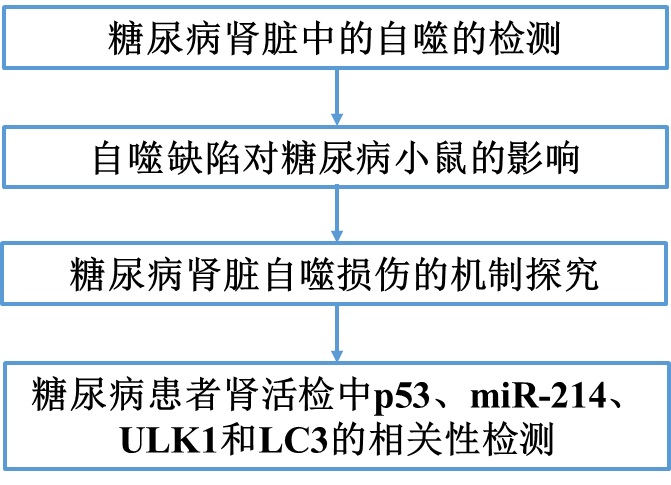
为了检测糖尿病肾脏中的自噬,我们首先测试了Akita 小鼠,一种胰岛素基因突变的1型糖尿病模型。Akita小鼠肾组织中LC3-I和LC3-II的水平明显低于野生型小鼠(图1A)。免疫组化染色显示,与WT小鼠相比,Akita小鼠肾小管细胞胞浆中LC3阳性的斑点较少(图1B)。电子显微镜还显示,与野生型小鼠肾脏相比,Akita小鼠肾脏的肾小管细胞中自噬小泡较少(图1C)。我们进一步监测了Akita小鼠糖尿病期间肾脏自噬变化的时间进程。7周龄时,Akita鼠和WT鼠的肾LC3表达均无差异(图1D)。LC3在Akita小鼠肾脏中的表达在第9周较低(图1E),但这种差异在第11周才具有统计学意义(图1F),表明糖尿病患者肾脏自噬随时间而减少。
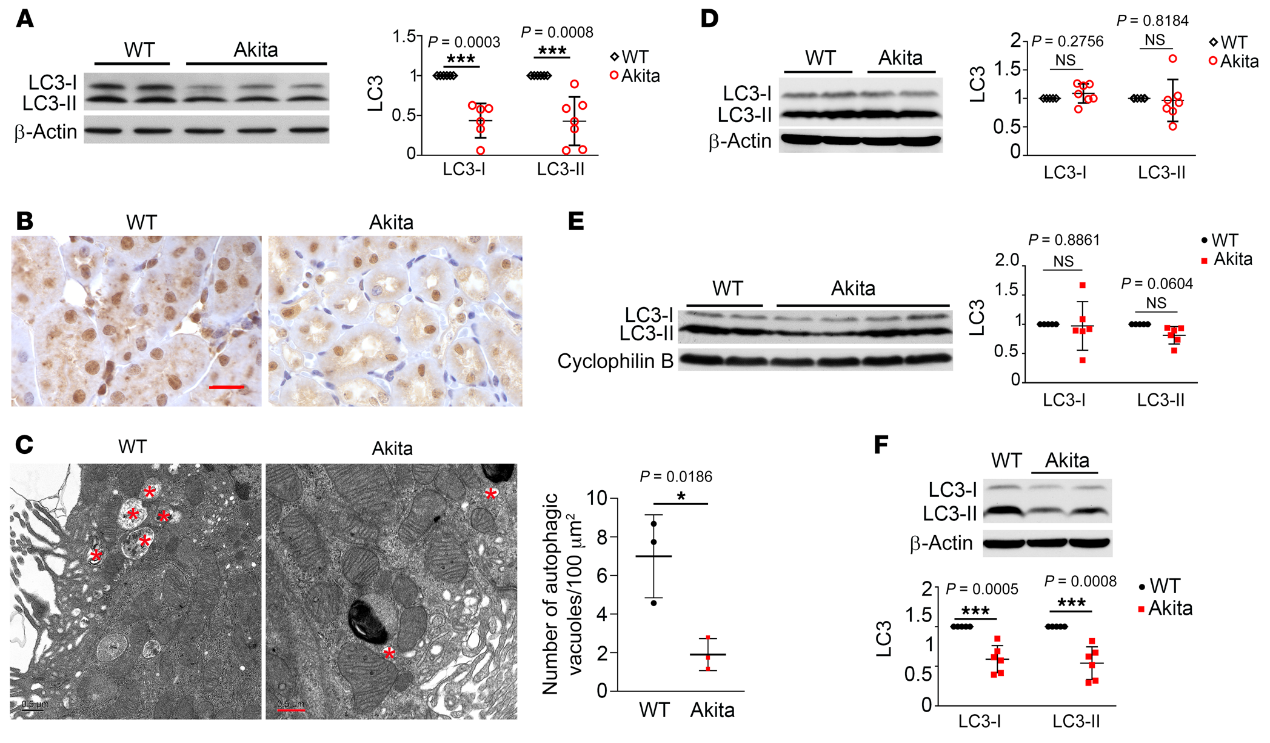
我们还检测了STZ诱导的C57BL/6小鼠糖尿病中的肾自噬。第11周,STZ处理的小鼠肾脏中LC3-I和LC3-II的表达水平明显低于对照组小鼠(图2A)。与未治疗的对照组小鼠相比,这些小鼠的肾小管细胞中的LC3点也较少(图2B)。
在糖尿病肾脏中观察到的自噬减少可能是由于自噬体形成和/或自溶体降解的变化。因此,我们通过使用CAG-RFP-GFP-LC3-转基因小鼠进一步监测自噬动力学,其中红色RFP与绿色GFP荧光的点状共定位指示自噬体,而没有GFP信号的仅RFP点状共定位指示自溶体。如图2C所示,与对照动物相比,STZ处理的小鼠肾小管细胞中的GFP- 和RFP-LC3 斑点明显较少,表明糖尿病肾的肾小管整体自噬受损。
2)近端小管的自噬缺陷扩大了糖尿病小鼠的肾脏肥大和组织损伤
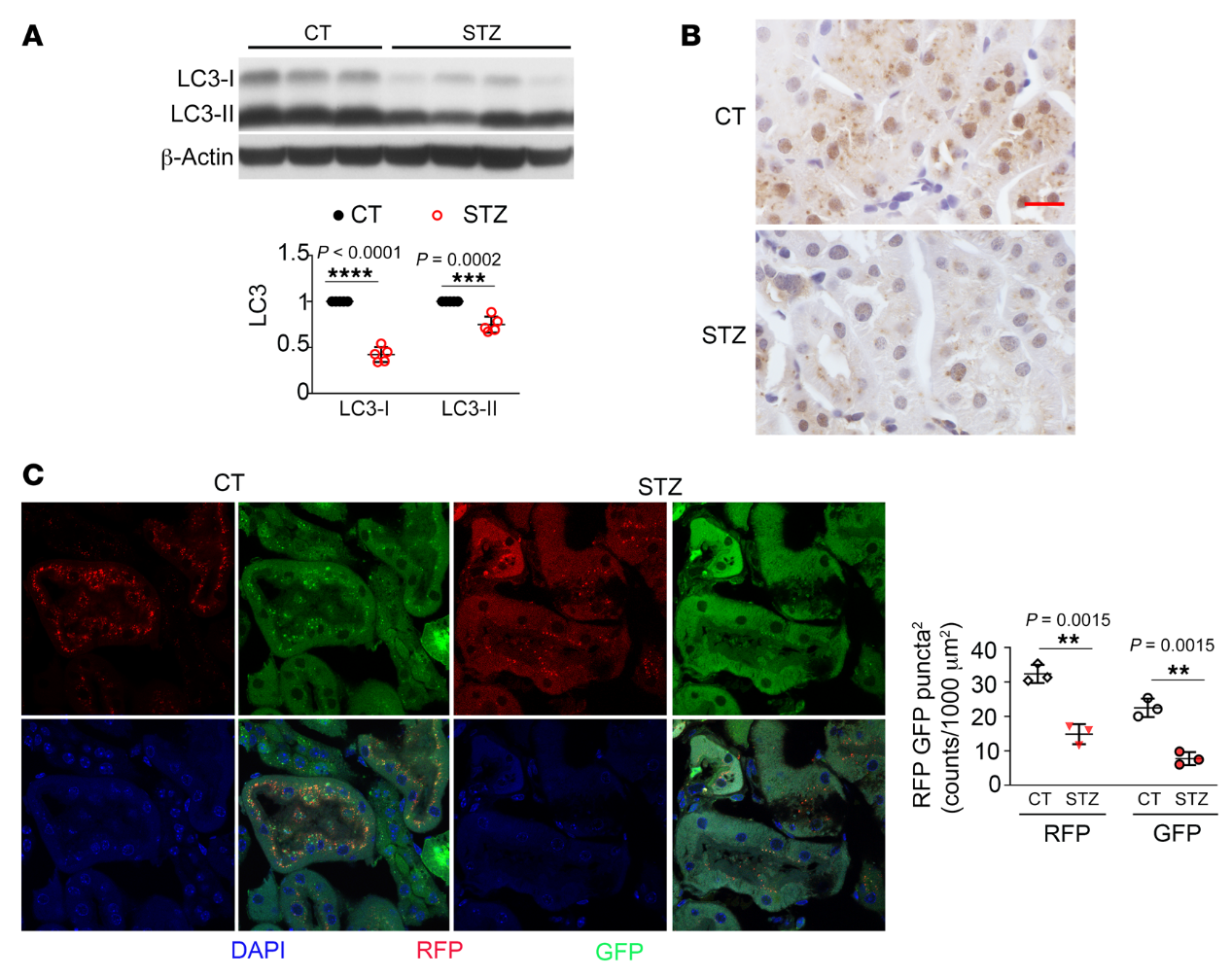
自噬是细胞分解代谢或降解的过程。因此,我们假设在糖尿病肾小管中观察到的自噬损伤可能导致肾脏肥大,这是DKD病的早期发病特征。为了验证这一点,我们首先确定了Akita小鼠肾脏近端小管自噬缺陷的影响。我们发现PT-Atg 7-/-Akita小鼠比PT-Atg7+/+ Akita小鼠具有更高的肾脏/体重比(图3A),这表明当肾小管自噬被抑制时,糖尿病肾脏会发生更严重的肥大。肾脏组织学分析显示,PT-Atg7-/-Akita小鼠的肾小管细胞嗜酸(图3B)。此外,PT-Atg7-/- Akita小鼠显示出肾小管损伤的其他迹象,包括肾小管细胞溶解和肾小管完整性丧失(图3B)。然后我们检查了巨噬细胞浸润。如图3C所示,我们在非糖尿病WT小鼠(不考虑Atg7表达)和Atg7表达的Akita鼠的肾组织中观察到很少的巨噬细胞。然而,PT-Atg7-/- Akita小鼠的肾间质中有大量巨噬细胞浸润,表明糖尿病期间近端小管的自噬缺陷促进了肾脏炎症。我们也用TUNEL染色检查肾细胞死亡。我们在PT-Atg7+/+ WT肾脏中未检测到TUNEL阳性细胞。在PT-Atg7–/–WT和PT-Atg7+/+Akita小鼠肾中观察到少量TUNEL阳性细胞,但在PT-Atg7-/- Akita小鼠肾中发现更多的TUNEL阳性细胞(图3D)。
3)肾小管自噬缺陷对DKD的长期影响。
用PAS染色,PT-Atg7-/- Akita小鼠比PT-Atg7+/+ Akita小鼠表现出更严重的肾小管损伤 (图4A)。此外,我们观察到PT-Atg7-/- Akita小鼠肾脏间质明显变宽(图4A)。同样,Masson’s trichrome染色检测到PT-Atg7-/- WT和PT-Atg7+/+ Akita小鼠肾间质中的边缘胶原沉积,这在PT-Atg7-/- Akita小鼠中显著增加(图4B)。PT-Atg 7-/- Akita小鼠的肾间质纤维连接蛋白染色也比其他组的小鼠多(图4C)。从功能上来说,与WT小鼠相比,PT-Atg7+/+和PT-Atg7-/- Akita小鼠的尿白蛋白/肌酐比(ACR)更高,但PT-Atg 7-/- Akita小鼠的ACR明显高于PT-Atg7+/+Akita小鼠(图4D),这表明肾小管自噬缺乏会加重糖尿病患者的肾功能衰退。总之,这些结果表明,肾脏近端小管的自噬缺陷可能会加重糖尿病患者的肾脏肥大和组织损伤,从而促进DKD病的进展。

4)糖尿病肾脏和高糖孵育的肾小管细胞中ULK1的下调
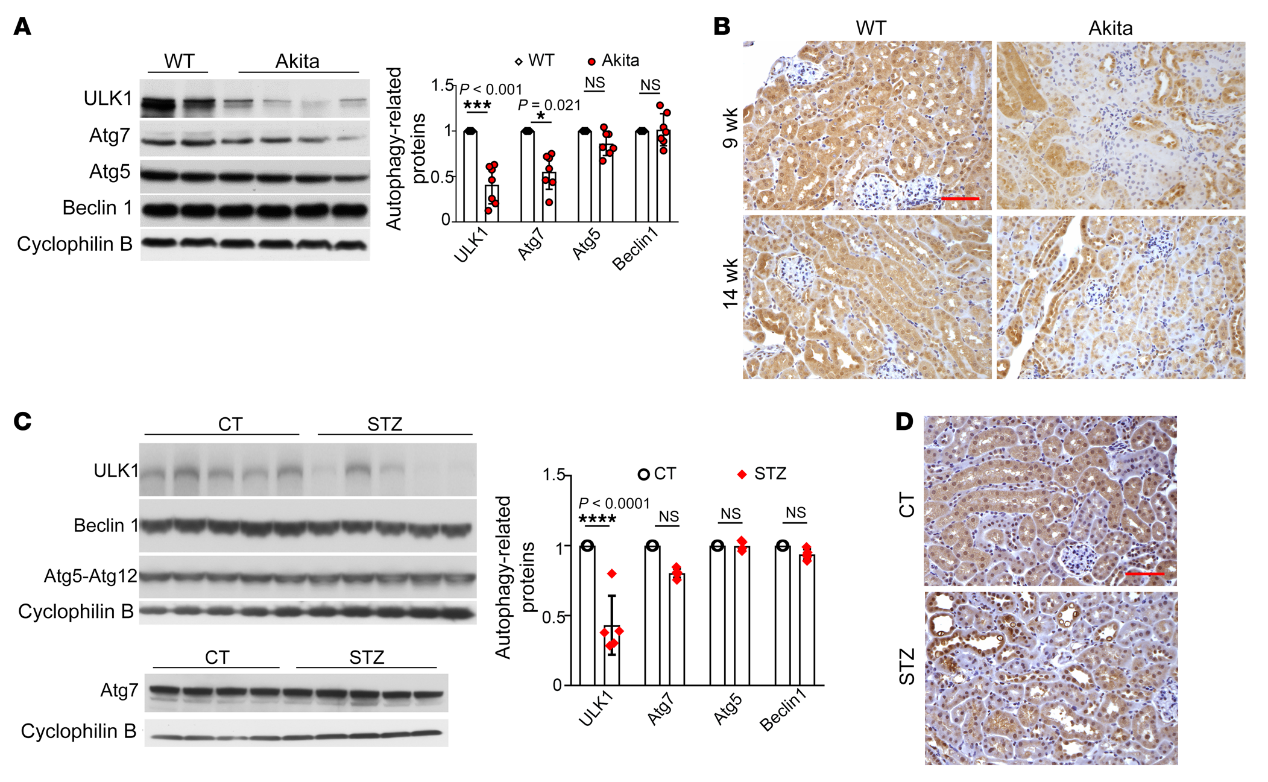
为了了解糖尿病肾脏自噬损伤的机制,我们首先通过免疫印迹法分析了Akita小鼠的关键自噬蛋白。我们发现糖尿病小鼠和它们的同窝仔在肾脏中表达相似水平的Beclin1和Atg5-Atg12复合物(图5A)。IHC染色进一步证实了糖尿病肾脏中ULK1的减少,尤其是在肾小管中(图5B)。类似地,在STZ诱导的糖尿病小鼠中,ULK1的肾表达减少(图5,C和D)。然而,STZ治疗没有改变小鼠肾脏中Atg7、Atg5或Beclin1的表达(图5C)。因此,糖尿病肾脏中一个显著的分子变化是ULK1的下调,这可能介导糖尿病中的自噬损伤。
5)在糖尿病肾脏中诱导miR-214抑制ULK1的自噬损伤、肾脏肥大和DKD
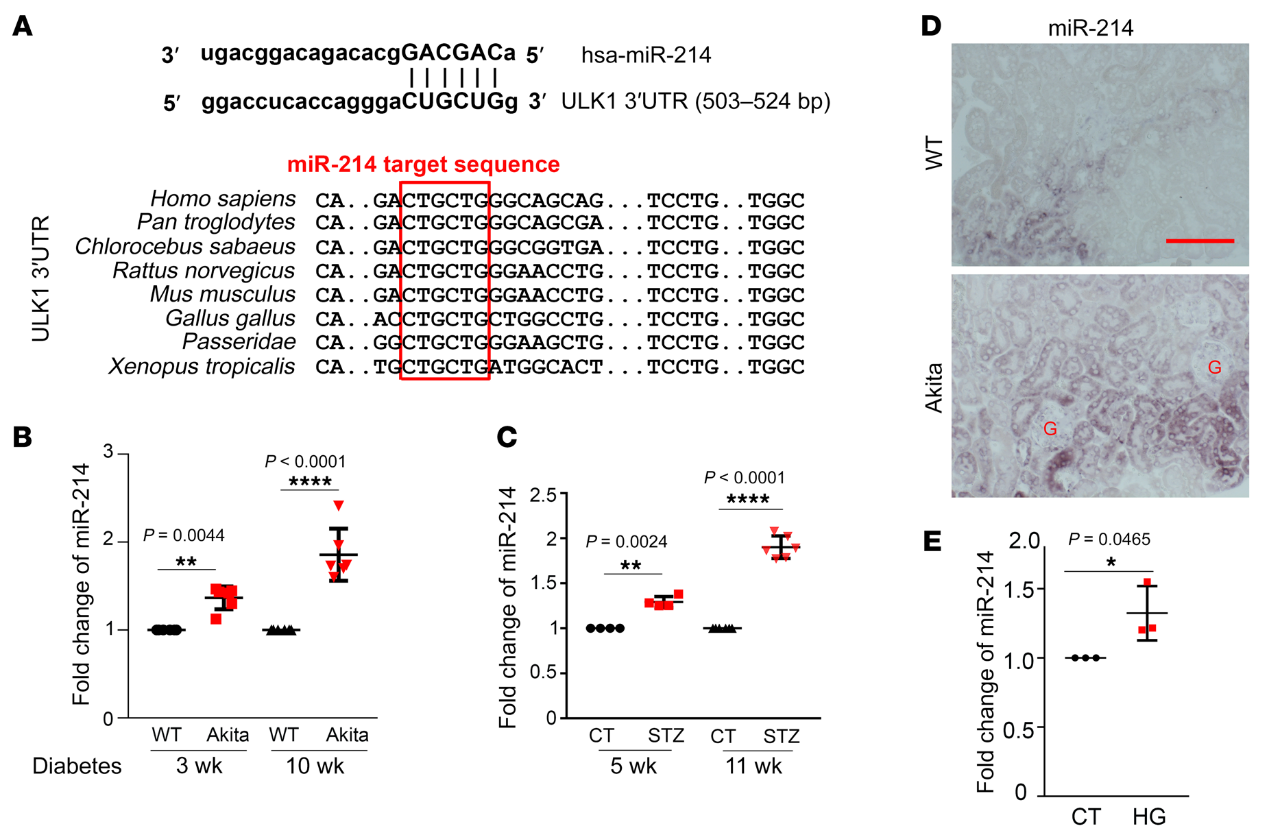
为了识别特定的microRNAs,我们首先使用miRanda microRNA目标预测数据库(http:/ /www.microrna. org)来预测可能以ULK1为目标的microRNAs。然后我们集中研究了那些与糖尿病和肾脏疾病有关的microRNAs。生物信息学分析表明miR-214是一种潜在的microRNAs,可以靶向糖尿病肾脏中的ULK1。我们在从热带爪蟾到人类的各种动物物种中鉴定了ULK1基因3’-UTR中的一个保守的miR-214靶向位点(图6A)。我们进一步检测到Akita小鼠和STZ诱导的糖尿病小鼠肾脏中miR-214的表达逐渐增加(图6,B和C)。使用ISH,我们进一步定位了糖尿病肾脏中miR-214的诱导,主要是在近端小管,而不是在肾小球(图6D),与非糖尿病WT组织相比,Akita小鼠肾脏中miR-214阳性管状细胞的数量显著增加。在体外RPTCs中,HG诱导miR214增加30%(图6E)。总之,这些结果证明了糖尿病期间肾小管细胞中miR-214的诱导,伴随着ULK1的下调。
为了确定ULK1是否是miR-214的真正靶标,我们首先将miR-214转染到人胚胎肾293 (HEK293)细胞中,这导致ULK1表达的显著降低(图7A)。相反, miR-214的抑制增强ULK1表达(图7B)。此外,抗miR214可防止HG诱导的ULK1(图7B)和LC3-II(图7C)表达的降低。我们通过荧光素酶报告分析,进一步确定ULK1是否是miR-214的直接靶点。如图7D所示,共转染miR-214降低了荧光素酶-ULK 1 3’-UTR转染细胞中的荧光素酶表达。值得注意的是,HG孵育也抑制了转染细胞中荧光素酶的表达,当miR-214的靶向序列在荧光素酶-ULK1 3’-UTR中突变时,这种作用部分减弱(图7E)。总之,这些数据表明ULK1是miR-214的直接靶标,miR-214可能抑制糖尿病中ULK1的表达。
为了证实anti–miR-214的结果,并进一步阐明miR-214在糖尿病肾脏中的致病作用,我们建立了近端小管特异性miR-214-敲除(PT-miR-214-/-)小鼠模型。PT–miR-214+/+ Akita小鼠肾脏中ULK1和LC3-II的表达降低,这两种蛋白在PT–MiR-214-/- Akita小鼠中被部分阻止 (图7,F和G)。此外,PT–miR-214-/-Akita小鼠的肾脏和肾脏重量/体重比小于PT–miR-214+/+ Akita小鼠,这表明miR-214有助于糖尿病患者的肾脏肥大(图7H)。值得注意的是,PT–miR-214-/- Akita小鼠的肾功能也明显更好(图7I)。

6)P53介导糖尿病肾脏中miR-214诱导的自噬损伤、肾脏肥大和DKD
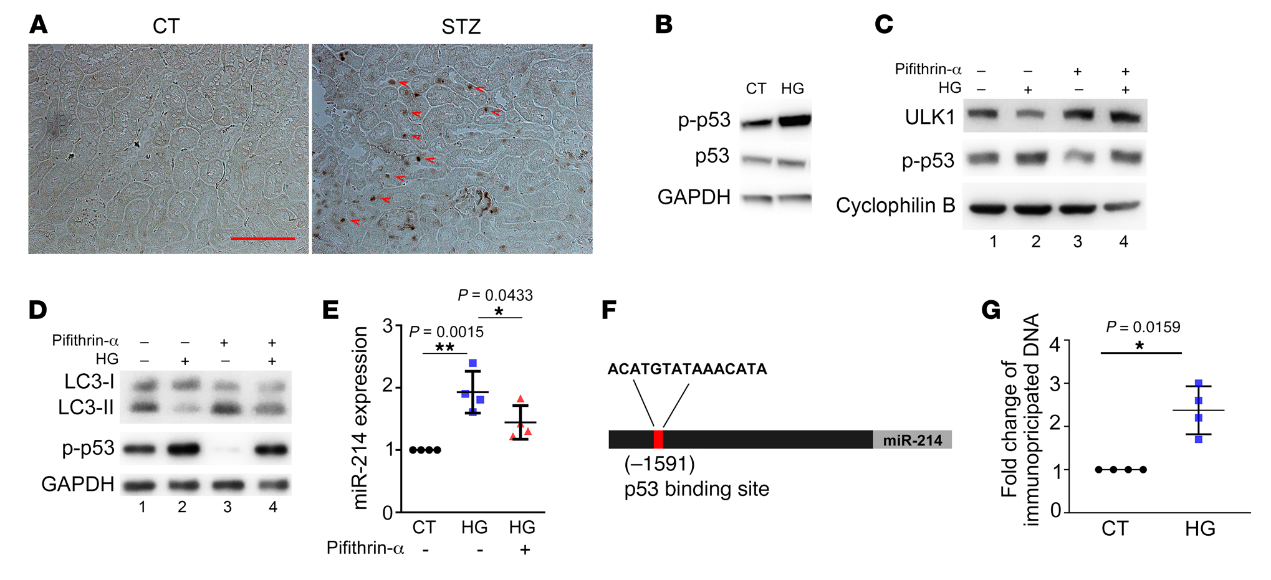
miR-214在DKD是如何诱导的?考虑到这个问题,我们筛选了几种可能的上游转录因子,其中p53在Akita和STZ治疗的小鼠肾脏中显著上调和激活(图8A)。在体外,RPTCs的HG孵育也诱导p53活化(图8B)。Pifithrin-α是p53的一种药理学抑制剂,可防止HG处理的RPTCs中ULK1和LC3-II表达的降低(图8,C和D)。值得注意的是,pifithrin-α还降低了HG处理细胞中miR-214的诱导(图8E)。这些结果表明p53可能介导肾小管细胞中miR-214的诱导以抑制糖尿病中的ULK1和自噬。
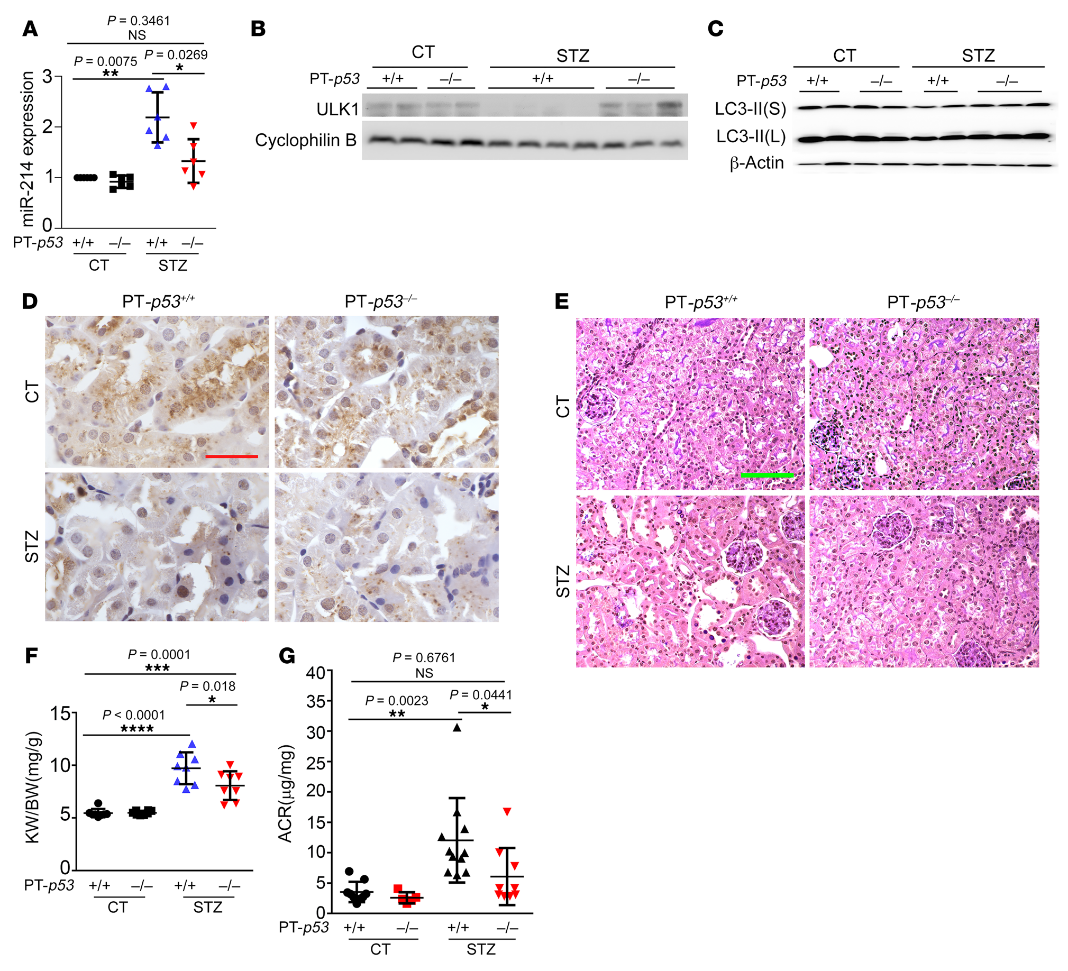
然后,我们使用JASPAR数据库(http:/ /jaspar.genereg.net/)分析miR-214基因的转录因子结合谱,并在其启动子区鉴定出一个假定的p53结合位点(图8F)。为了验证p53与该位点的结合,我们进行了ChIP分析以检测抗p53免疫沉淀中的结合位点序列。如图8G所示,HG孵育诱导p53与miR-214基因启动子序列的结合增加了2.3倍,该序列具有假定的结合位点。为了阐明p53是否在体内调节miR-214,我们使用了我们以前工作中的近端小管特异性p53敲除(PT-p53–/–)小鼠模型。PT-p53-/-小鼠和它们的PT-p53+/+同窝小鼠用STZ治疗以诱发糖尿病。经STZ治疗后,PT-p53+/+小鼠肾脏中miR-214的诱导作用显著,而PT-p53-/-小鼠中miR-214的诱导作用减弱(图9A)。与此同时,糖尿病相关的ULK1和LC3-II表达的降低在PT-p53-/-小鼠中被抑制(图9,B和C)。在STZ诱导糖尿病后,PT-p53-/-肾在肾小管中也比PT-p53+/+肾有更多的LC3阳性点或自噬体(图9D)。在PAS染色中,糖尿病性PT-p53+/+小鼠表现出明显的肾脏组织病理学变化,包括肾小管扩张、刷状缘丧失和肾小管萎缩,这些在糖尿病性PT-p53-/-小鼠中显著减少(图9E)。糖尿病期间肾脏重量和肾脏重量/体重比增加,但PT-p53-/-小鼠的增加被部分抑制,表明这些动物的肾脏肥大减少(图9F)。值得注意的是,在STZ诱导的糖尿病中,PT-p53-/-小鼠的ACR显著低于PT-p53+/+小鼠(图9G),表明肾功能更好。综上所述,这些结果表明糖尿病中p53激活以诱导miR-214,miR-214随后抑制ULK1的表达,导致自噬损伤、肾肥大和DKD。
7)糖尿病患者肾活检中p53、miR-214、ULK1和LC3的相关性
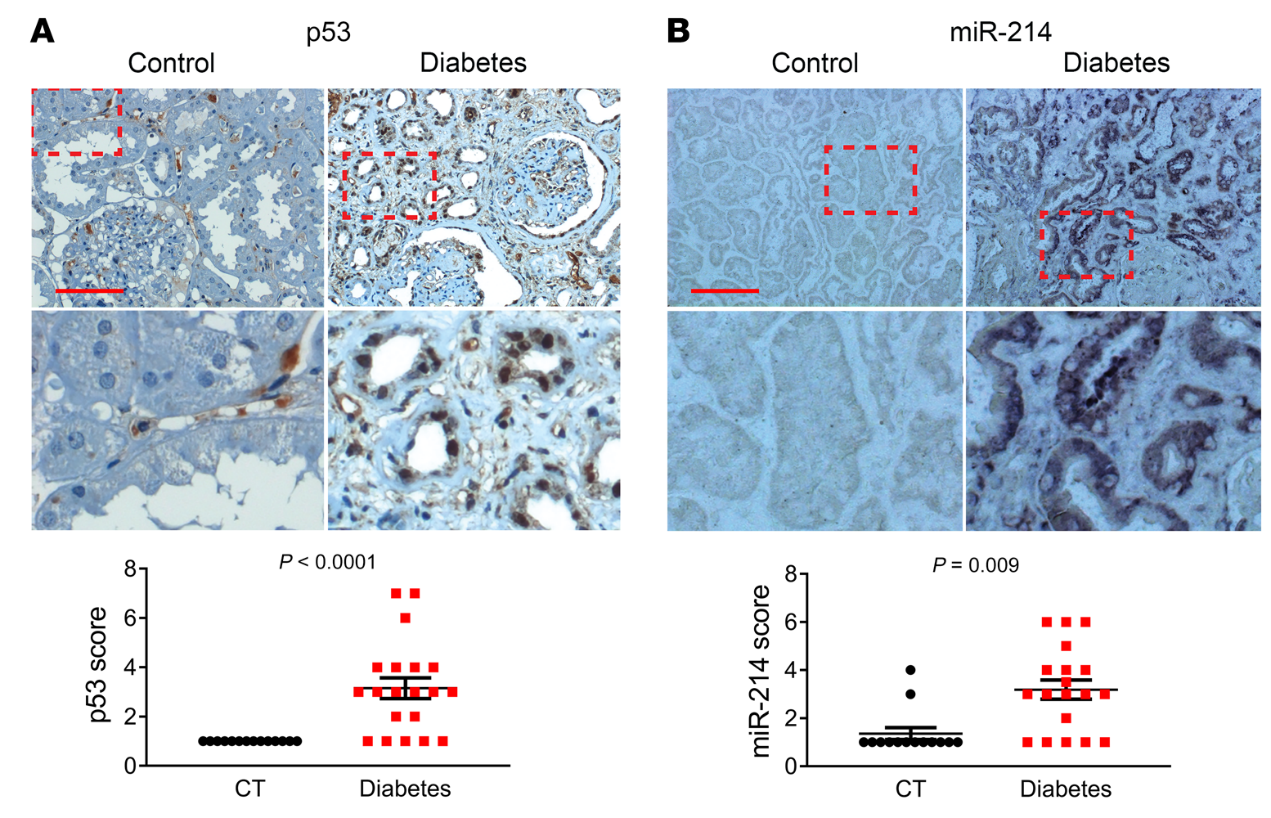
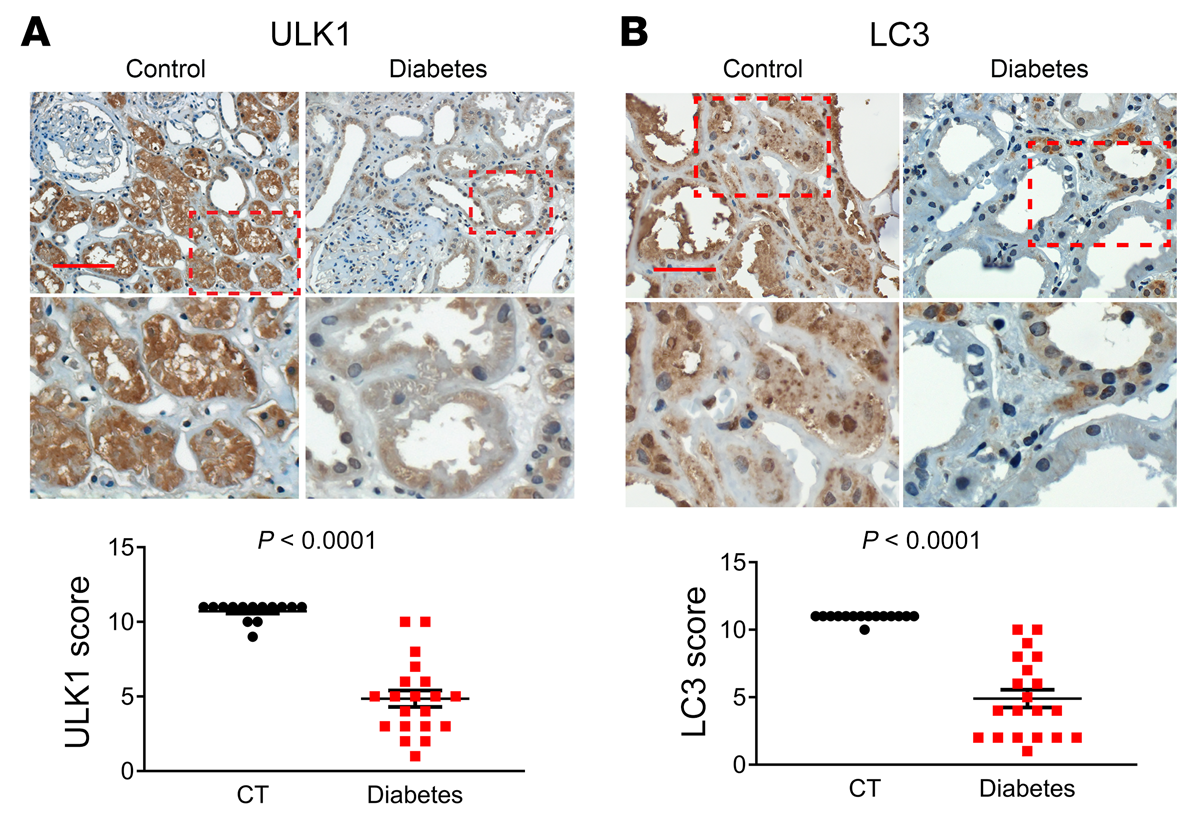
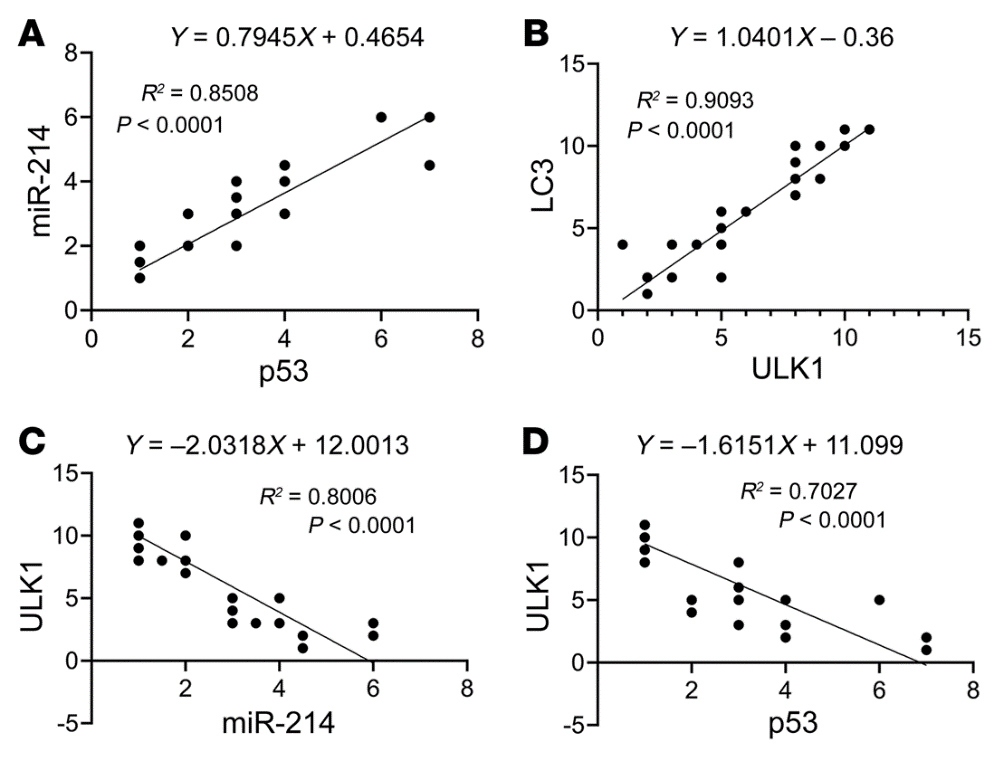
我们的动物和细胞模型结果揭示了p53/miR-214/ULK1途径,该途径导致自噬缺陷、肾脏肥大和糖尿病肾功能下降。为了确定这些发现的临床相关性,我们检测了糖尿病患者肾活检中p53、miR-214、ULK1和LC3的表达。在IHC分析中,没有一个对照肾活检标本p53染色阳性,而来自糖尿病患者的20个肾活检标本中有15个显示p53染色(图10A)。值得注意的是,在糖尿病肾脏样本中,p53主要在扩张肾小管的细胞核中表达。ISH显示,14例对照肾活检中只有2例miR-214染色阳性,而糖尿病患者20例肾活检中有15例阳性(图10B)。有趣的是,miR-214在肾小管中以p53的形式存在,但主要在细胞质中。相反,对照肾活检中的大多数肾小管对ULK1染色强烈,而糖尿病肾活检中的许多肾小管对ULK1染色较低(图11A)。与对照活检相比,糖尿病患者的肾活检也显示明显较少的LC3点或自噬体(图11B)。此外,线性回归分析显示糖尿病肾活检中p53和miR-214表达之间(图12A)以及ULK1和LC3表达之间(图12B)存在显著正相关。相比之下,我们注意到miR-214和ULK1的表达之间(图12C)以及p53和ULK1的表达之间(图12D)存在显著的负相关。这些结果提供了进一步支持p53/miR-214/ULK1轴在糖尿病自噬损伤中的作用,并建立其临床相关性。
结论:我们在DKD的实验模型和糖尿病患者的肾脏中都证实了肾小管细胞的自噬功能障碍。自噬损伤有助于肾脏肥厚,相关病理,以及DKD的进展。机制上,我们发现p53诱导了DKD中的miR-214,抑制了自噬启动的关键蛋白激酶ULK1,导致自噬损伤。